Author(s): Kirubel Zemedkun Gebreselassie*, Amine Bourbia, Logan Minshew and Eden Z Gebreselassie
A 40-year-old male with past medical history of chronic pain on methadone for a few years, hypertension, and obesity presented with worsening dyspnea and was found to have significant hypoxemia. A CT-angiogram of the chest showed extensive diffuse bilateral pulmonary infiltrates with bilateral hilar and mediastinal lymphadenopathy. Extensive work-up exploring infectious and rheumatologic etiologies was unremarkable. Patient was empirically treated with IV antibiotics, high dose steroids and aggressive diuresis without significant clinical improvement. Further more, serial chest radiographs showed no improvement. Right side heart catheterization revealed normal pulmonary wedge pressure with elevated right-side pressure consistent with pre-capillary pulmonary hypertension. Right video assisted thoracoscopic surgery (VATS) wedge lung biopsy was done and tissue biopsy revealed marked intra-alveolar congestion by red blood cells, patchy aggregates of pulmonary macrophages within these alveolar spaces, and thickened alveolar septa. The thickened alveolar septa contained an increased number of dilated capillaries and mild numbers of chronic inflammatory cells. Alveolar macrophages contain anthracotic pigment as well as occasional pigment compatible with hemosiderin. There was no evidence of significant fibrotic change, fibroblastic foci, granulomas, or other significant inflammatory changes. This is consistent with capillary hemangiomatosis like change consistent with chronic venous hypertension.
Pulmonary veno-occlusive disease (PVOD), also called pulmonary capillary hemangiomatosis (PCH) is a rare condition that refers to group 1 pulmonary hypertension WHO classification [1]. The true incidence of PVOD is not well-known because of misclassification to idiopathic pulmonary arterial hypertension and chronic pulmonary thromboembolism [2]. Annual incidence of PVOD/ PCH is estimated to be 0.1 to 0.2 cases per million persons in the general population making our case very rare and unique [3]. We are presenting a 40-year-old male patient who presented to our facility with one month of progressive shortness of breath with significant hypoxemia, and extensive diffuse bilateral pulmonary infiltrates with bilateral hilar and mediastinal lymphadenopathy. The differential diagnosis for such lesions is extensive including infectious etiology, pulmonary hemorrhage, interstitial lung disease, autoimmune disease, side effects of drugs and toxins, and even malignancy [4]. Detailed work-up for infectious and rheumatologic diseases was nonremarkable. Establishment of diagnosis and treatment of PVOD/PCH has been a challenge in the clinical practice of pulmonary hypertension. Although the definite diagnosis is based on pathological findings, it is necessary to make an early clinical diagnosis considering the poor prognosis of PVOD/PCH [5].
This is a 40 years old male with past medical history of chronic pain on methadone for several years, essential hypertension, and obesity who presented with worsening of shortness of breath. The patient reports he has been short of breath since his prior admission a month ago when he was admitted for acute kidney injury. His kidney malfunction was presumed to be of prerenal causes due tomultiple rounds of nausea and vomiting over the course of several days. He was given IV fluids and his kidney function returned to baseline and was discharged home in good condition after a four-day hospital stay. CT abdomen and pelvis without contrast that was done for renal failure work-up showed diffuse ground glass attenuation and some coarse interstitial markings on the lower lung base window. Radiologic concern was for pulmonary edema versus pulmonary fibrosis. His oxygen requirement was only 2 L/min nasal cannula oxygen that was later weaned off.
The patient returned to the hospital after a month, for shortness of breath that never improved since his discharge. His shortness of breath progressed over the past 24 hours before admission which prompted him to come to the ER. He was found to be in respiratory distress requiring 100% FiO2 BiPAP. Chest x-ray showed bilateral interstitial and patchy alveolar opacities suggesting possible multifocal pneumonia. There was no evidence for pulmonary thromboembolism on chest CT-angiogram. However, extensive diffuse bilateral pulmonary infiltrates with bilateral hilar and mediastinal lymphadenopathy with subcarinal nodes measuring 5.0 x 3.2 cm in diameter in the right paratracheal lymph node conglomerate measuring up to 3.7 x 4.3 cm with other numerous enlarged nodes were also noted (Figures 1 & 2).
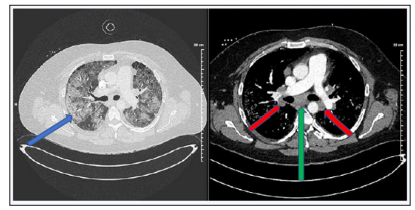
Figures 1 & 2: Extensive diffuse bilateral pulmonary infiltrates (blue arrow) which can be seen in multifocal pneumonitis, ARDS, pulmonary hemorrhage, etc. Without evidence of pleural or pericardial effusion.
Bilateral hilar (green arrow) and mediastinal lymphadenopathy (red arrows) with subcarinal nodes measuring 5.0 x 3.2 cm in diameter, and a right paratracheal lymph node conglomerate measuring up to 3.7 x 4.3 cm. Numerous other enlarged nodes are present as well.
Blood gas was consistent with severe ARDS, that required high flow nasal cannula oxygen treatment (WHAT WAS THE PF RATIO). The diffuse bilateral infiltrates with prominent hilar mediastinal lymphadenopathy seen on chest CT angiogram appeared to be progressed in comparison to lung base view on CT abdomen/pelvis a month prior. Infectious work up including viral respiratory panel, COVID-19, acute hepatitis panel, HIV, and procalcitonin, Fungitell, Aspergillus Ag, and Histoplasma urine Ag were all negative and the patient was started on ceftraizone and azithromycin empirically with minimal improvement. He was also treated with diuresis for presumed pulmonary edema with no improvement. Of note, his echocardiogram revealed an ejection fraction of 55-60% with normal left ventricular diastolic function and he had normal kidney function at this time. He denied history of vaping or cocaine use. He smoked half a pack of cigarettes daily for eight years and stopped smoking six months prior to his first admission. Although bronchoscopy would be of benefit to rule out other infectious etiologies, the patient was at high risk of prolonged ventilatory support post procedure, and was deferred unless there was further decline in the patient’s respiratory status to the point of intubation.
Rheumatologic work-up showed elevated C-reactive protein and sedimentation rate. An ANA was negative, C3 levels were elevated, and C4 levels were normal. Both myeloperoxidase and proteinase-3 ANCA antibodies were also negative. There is no personal or family history of autoimmune disease. The clinical presentation was somewhat challenging, with extensive and broad differential diagnosis including but not limited to, organizing pneumonia, acute or chronic eosinophilic pneumonia and even sarcoidosis. The patient was started on systemic steroids with methylprednisolone IV 125 mg every 6 hours with hopes of improving respiratory status and deferring intubation and mechanical ventilation. Our patient appeared to be responding to steroid therapy, and treatment continued at tapered doses and later changed to oral prednisone. It was felt that he would need a prolonged course of steroid treatment and hence trimethoprim/ sulfamethoxazole prophylaxis started.
The patient’s respiratory status unfortunately worsened again in the subsequent days requiring increased amount of oxygen support. Antibiotics were discontinued, and aggressive diuresis was reinitiated despite persistently low levels of NT-proBNP and normal echocardiographic findings. Cardiac catheterization was planned after optimal diuresis to look for underlying left side heart failure and assess response to treatment. CT-angiogram of the chest was repeated showing extensive mediastinal and hilar adenopathy which was slightly improved compared to previous study. Lungs showed diffuse airspace disease bilaterally that was less dense but slightly more diffuse than the previous study (Figure 3 & 4).
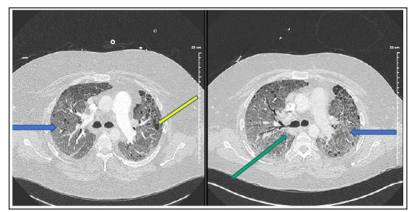
Figures 3 & 4: Extensive mediastinal and hilar adenopathy (green arrow) is again noted which may be slightly improved as compared to prior study. Lungs again show diffuse airspace disease bilaterally which is somewhat less dense overall but slightly more diffuse than on prior. This appearance is overall nonspecific. Underlying cystic lung disease is also noted (yellow arrow).
Patchy ground glass interstitial opacities (blue arrow) throughout both lungs that are suspicious for an infectious or inflammatory process remain unchanged. Enlarged mediastinal, right hilar lymph nodes, and shotty mediastinal lymph nodes that are most likely reactive are unchanged. Small right pleural effusion is new.
Underlying cystic lung disease is also noted. The decision was made to proceed with wedge lung biopsy once oxygen requirements dropped to an acceptable range. In mid-May he had a right VATs procedure for lung biopsy and the specimen was sent to an outside tertiary facility. Microscopic sections of the wedge demonstrated marked intra-alveolar congestion by red blood cells, patchy aggregates of pulmonary macrophages within these alveolar spaces, and thickened alveolar septa (Figures 5 & 6).
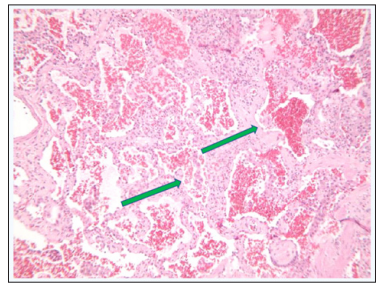
Figure 5: Diffuse Thickening of Alveolar Septae (green arrow)
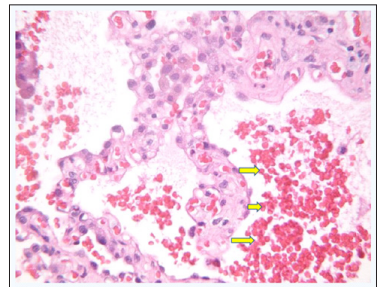
Figure 6: Proliferation of Capillary-Like Vessels within Alveolar Septae (yellow arrow)
The thickened alveolar septa contained an increased number of dilated capillaries and mild numbers of chronic inflammatory cells. Alveolar macrophages contain anthracotic pigment as well as occasional pigment compatible with hemosiderin (Figure 7).
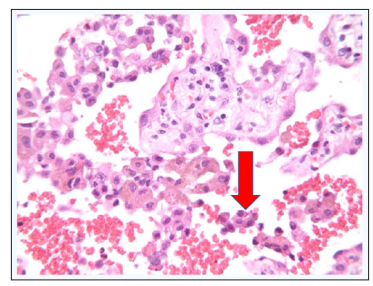
Figure 7: Hemosiderin Laden Macrophages (red arrow)
There was no evidence of significant fibrotic change, fibroblastic foci, granulomas, or other significant inflammatory changes. Final diagnosis was reported as capillary hemangiomatosis like change consistent with chronic venous hypertension.
Top differential diagnoses based on pathology results were venous hypertension from a cardiac cause, chronic thromboembolism, and/ or obstructive causes in the mediastinum need to be considered. In the absence of these, pulmonary vaso-occlusive disease could be the top differential diagnosis. Patient underwent a right heart catheterization. He was found to have elevated mean pulmonary arterial pressure of 34 mmHg, right ventricular systolic pressure of 42 mmHg, and mildly increased pulmonary vascular resistance of 244 dynes/sec/cm-5 consistent with precapillary pulmonary hypertension. His mean pulmonary capillary wedge pressure was normal at 8 mmHg. Echocardiogram showed normal ejection fraction with a dilated right heart and preserved right ventricular systolic function. No significant valvular abnormalities were identified by doppler. Negative saline contrast study ruled out left to right shunting. Patient had transient peripheral eosinophilia with normal immunoglobin (Ig)E levels. No sites of ventilation/ perfusion mismatch were identified on ventilation/perfusion scintigraphy with a low probability of pulmonary embolism. Some interstitial thickening was noted on VQ scan. Right heart catheterization results in context of the lung biopsy findings were highly suggested of PVOD/PCH. Transfer of the patient to a tertiary care center for evaluation of lung transplant was initiated. The patient was started on sildenafil for pulmonary hypertension in the meantime with which he had clinical improvement and was able to wean down oxygen requirements to 6 L/min nasal cannula and he was discharged after three and half months of hospital stay. However, he returned in a few days, due to worsening pulmonary edema and increased oxygen requirements.
It is likely that PVOD is a result of a multiplicity of factors including infection, genetic predisposition with reported disease prevalence amongst siblings, toxin exposure, thrombotic diathesis and autoimmune disorders. Although there is no concrete data linking a specific infection to PVOD, it is reported that Toxoplasma gondii, measles, Epstein-Barr, cytomegalovirus, and HIV are possible causes. Chemical exposures have been linked to a number of cases of PVOD as well [6]. Early cases described an association between ingestion and inhalation of powdered cleaning products that contain silica, soda ash, dodecyl benzyl sulfonate, and trichloro-s-triazinetriome. Additionally, PVOD became a well-recognized complication of antineoplastic chemotherapy such as bleomycin. Concerning thrombotic diathesis, several reports described the increased platelet adhesiveness in PVOD and the condition arising in hyper coagulable patients as in pregnancy or oral contraceptive use [6].
The differential diagnosis of diffuse parenchymal lung disease is extensive and determining the tempo of the pathological process helps to narrow down the most likely etiology [7]. Acute processes less than 4 to 6 weeks in duration most commonly include infectious processes, pulmonary edema both cardiogenic and noncardiogenic, pulmonary hemorrhage, or aspiration [8]. In addition, some interstitial lung disease may present acutely such as hypersensitivity pneumonitis, acute eosinophilic pneumonia, druginduced lung disease, acute interstitial pneumonia, and cryptogenic organizing pneumonia/idiopathic bronchiolitis obliterans with organizing pneumonia. [9,10]. Our patient’s clinical presentation can rule out most of the chronic lung diseases given less than 4 weeks since symptom onset. The extensive negative workup for infectious disease and lack of response to antibiotic treatment makes bacterial and atypical infections less likely. The diagnosis of cryptogenic organizing pneumonia (COP) depends upon demonstration of the typical histopathologic features of excessive proliferation or“;plugs”; of granulation tissue within alveolar ducts and alveoli associated with chronic inflammation in the surrounding alveoli is not seen in our patient [11]. Pulmonary renal syndromes are a possible differential diagnosis that usually present with a rapidly progressive glomerulonephritis picture and alveolar hemorrhage. The majority of cases of pulmonary-renal syndrome however are associated with ANCAs, either c-ANCA or p-ANCA, due to autoantibodies against the target antigens proteinase-3 and myeloperoxidase respectively [12]. The lack of improvement on high-dose steroid treatment also makes most of the inflammatory and autoimmune diseases such as sarcoidosis less likely.
Most of the idiopathic interstitial pneumonias are chronic and UIP is one of the common forms which typically presents in the sixth or seventh decade of life [13]. The absence of fibrosis on pathology and clinical picture does not support a diagnosis of UIP in our patient. One of the interstitial lung diseases associated with smoking is desquamative interstitial pneumonitis with the most striking feature on pathology being the presence of numerous mononuclear cells with minimal or mild associated fibrosis [14]. Acute interstitial pneumonia (AIP) presents with rapidly progressive respiratory failure analogous to acute respiratory distress syndrome similar to our patient. In addition, one may find alveolar septal dramatically thickened, however the main finding is extensive interstitial fibroblastic proliferation with an edematous appearing stroma [15]. He had been taking methadone for more than 10 years, and reports of noncardiogenic pulmonary edema with methadone have been reported, however it usually develops rapidly within the first few hours following injection, but rarely occurs as late as 24 hours [16].
The apparent precapillary pulmonary hypertension pattern on right heart cardiac catheterization, together with consistent symptoms, radiologic finding, and absence of other differential diagnosis highly suggest the diagnosis of PVOD/PAH in our patient. Progressive dyspnea and fatigue are the principal clinical manifestations of both PVOD and PCH [17]. Pathology remains the most definitive modality to diagnose PCH [18]. Histologic differential diagnosis includes pulmonary venoocclusive disease, acute vascular congestion, and atelectasis artifact. PVOD may show venous occlusion; however, the distinction between PVOD and PCH may be arbitrary [19]. Acute pulmonary congestion may at first resemble PCH, but lacks the proliferation of capillarylike vessels seen in PCH. Atelectasis artifact will also not show capillary-like vascular proliferation in alveolar septae. Most cases of PVOD/PAH are idiopathic and it has been postulated that it may represent a common aberrant response to an inciting endothelial injury that leads to widespread fibrosis of pulmonary venules [20]. The diagnostic sensitivity of the classic PVOD triad of patchy ground glass opacities, septal lines, and mediastinal adenopathy is greater than 80 percent [21]. The clinical diagnosis of PVOD can be made in most patients using a constellation of clinical, computed tomographic (contrast-enhanced CT), physiologic, and hemodynamic findings of right heart cardiac catheterization [22]. Delays in the diagnosis of PVOD are common since the condition is rare, the presentation is nonspecific, and not every patient presents with classic features. Many patients are initially presumed to have heart failure, CTEPH, or parenchymal lung disease, such as sarcoidosis, cystic fibrosis, or pneumoconiosis (due to chronic interstitial changes on chest radiographs) [23]. Prognosis remains poor with median survival of 3 years [24]. The reason for our patient’s third admission could be due to development of pulmonary edema related to recently started vasodilator treatment which is a well reported complication with PAH targeted therapies in patient with PVOD/PCH [25].
In conclusion, PVOD remains a poorly understood syndrome. It is a disease that is likely a result of multiple insults of diagnostic importance due to its poor outcomes and therefore rapid evaluation for the purpose of early enlistment for transplantation. Furthermore, additional research is required to precisely define the risk factors associated with this condition and to determine optimal therapy.
Our acknowledgment goes to Dr. Christopher Kligora (Atrium Health Floyd Pathology Department) who provided us with the pathology slides and Dr John Whalen who helped with the editing of our paper.