Author(s): Sowmya Ramanan*, Shivang Saxena and Baiju S Dharan
Isolated congenital pulmonary valve regurgitation (IPVR) is very rare. Absent/ bicuspid/ dysplastic pulmonary valves are the common congenital causes of this condition. Here we present a rare cause of IPVR caused by absent single pulmonary cusp. A review of literature to establish timing and management of isolated pulmonary regurgitation is also attempted
Isolated pulmonary valve regurgitation (IPVR) is very rare (0.8% of CHD) [1]. We present an even rarer case of IPVR caused by the absence of a single pulmonary cusp. Guidelines are not established regarding indications and timing of valve replacement for congenital IPVR. With review of literature, we propose a management outline for this condition.
15-year-old boy, with a diagnosis of congenital dysplastic pulmonary valve (PV), presented with a one-year history of palpitation and breathlessness (NYHA class II). He had sinus tachycardia and grade 4/6 early diastolic murmur at the left 2nd intercostal space. Electrocardiogram showed sinus rhythm, right axis deviation with complete right bundle branch block. Chest X-ray showed increased cardiothoracic ratio with dilated main pulmonary artery (MPA). Transthoracic echocardiography (TTE) showed paradoxical septal movement; dilated PV annulus (31mm), with MPA (57 mm) and branch pulmonary artery (PA) measurements of right (24mm) and left (26mm),and severe PR. Cardiac computed tomogram (CT) and magnetic resonance imaging (MRI) were done to delineate pulmonary artery anatomy and right ventricular (RV) dimensions. Imaging revealed severe PR with absence of single PV leaflet and dysplastic but normal sized remaining 2 leaflets (Figure 1 ). RV ejection fraction (RVEF) was 48% and RV end diastolic volume index (RVEDI) was 187ml/m2. There was no bronchial compression from dilated PAs.
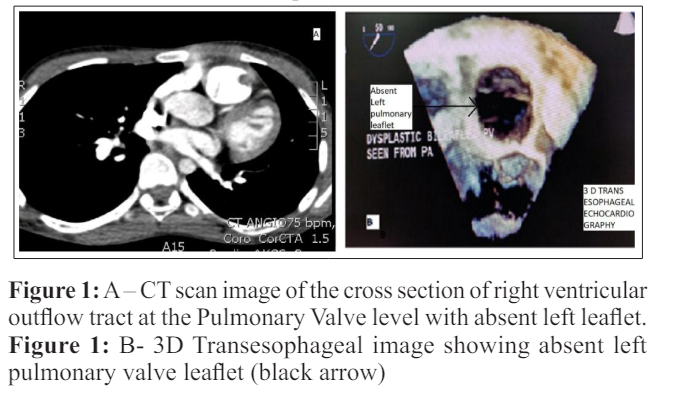
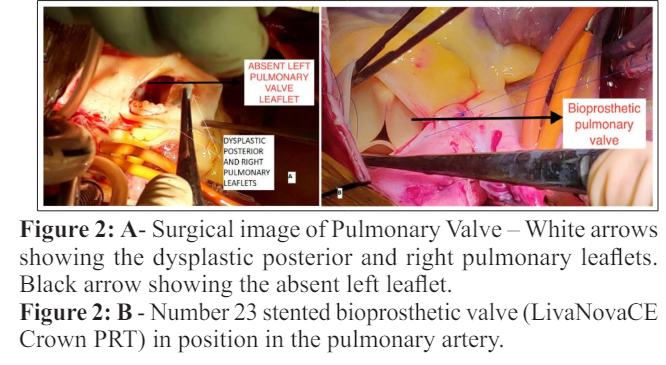
At surgery, MPA was dilated with dilated branch PAs. Posterior and right pulmonary cusps were dysplastic and left cusp was absent. (Figure 2). Number 23 stented bioprosthetic valve (LivaNovaCE Crown PRT) was used for PV replacement. Postoperative period was uneventful.
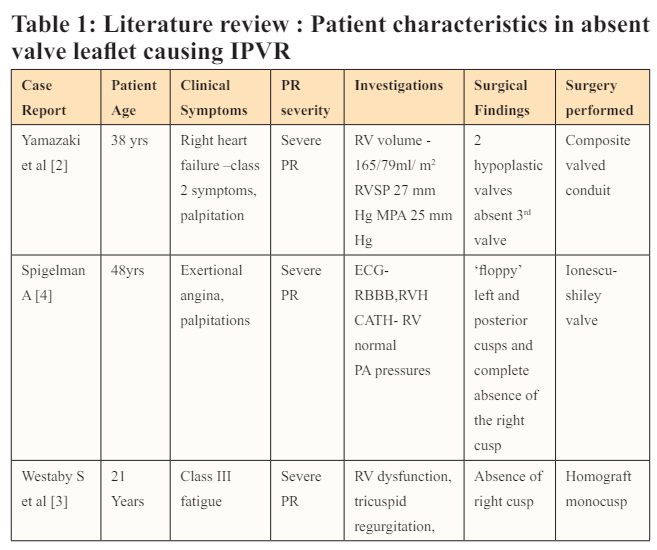
Among the causes for congenital IPVR, idiopathic dilatation of MPA is most common (0.6% of CHD) and bicuspid PVs are considered to be the rarest (0.2% of CHD) [1]. Bicuspid PV refers to an anatomy similar to bicuspid aortic valves, wherein the valve is bi-commissural. In a semilunar valve with absent single cusp, there are 2 dysplastic or normal cusps attached at a single commissure and the third cusp is absent. There are only 3 reported cases with absent single cusp causing severe PR (Table 1) [2,3]. Ours is the fourth.
IPVR is usually well tolerated. Symptoms develop around 4th decade of life when PR becomes chronic and RV dysfunction established. Irrespective of etiology, symptoms are those of right heart failure, arrhythmias, syncope and sudden cardiac death [1]. While MPA and branch PA dilatation is a noted pathology, reports of airway compression by these dilated vessels is scarce. Literature search revealed analysis of this observation only in a subgroup of infants with prenatal diagnosis of absent PV syndrome; those with associated Tetralogy of Fallot (TOF) and with intact ventricular septum (which would later become IPVR) [4]. The observations from these rare reports (2/15 cases) are that presence of a ductus arteriosus to the MPA decompresses the branch PA and thereby reduce chances of airway compression. Long term outcomes of this cohort are unavailable.
Due to rarity of cases, literature is devoid of guidelines for operating on cases with congenital IPVR [5]. Azam Ansari has reported 69 cases since 1955 and has compared the etiology and management of this rare lesion [1]. In the current era, the proposed investigations such as fluoroscopy, cardiac catheterization and indicator dilution studies would be replaced by echocardiography, CT scan and MRI. With newer investigations, the indications of surgery also change. 2D TTE with color flow Doppler would be the recommended first line investigation for establishing diagnosis and severity of the PR [6]. Manivarmane et al recommended routine use of biplane and 3D imaging in both TTE and transesophageal echocardiography to delineate PV anatomy in IPVR [6]. The use of 3D transesophageal echocardiography is currently limited to older patients because of technical limitations.
Cardiac MRI would be essential in planning management. Since volumetric data of IPVR is scarce, planning treatment protocols require extrapolation from the published literature on corrected Tetralogy of Fallot. Parameters considered are Effective Regurgitant Orifice Area, Regurgitant volumes, RVEF, RVEDI and RV end systolic volume index (RVESI) [7].
Therrien et al reported that PV replacement fails to normalize RV dimensions when performed for RVEDI ≥170ml/m2 and RVESI ≥ 85 ml/m2 in corrected Tetralogy of Fallot. The average duration between surgeries in this study was 29+/- 9 years. The indications of surgery were the presence of RV dilation and hypokinesia (100%), severe PR (94%), exertional dyspnea (70%), and recurrent or sustained arrhythmias (59%). Multi planar MRI imaging was considered better than echocardiography and radionucleotide imaging for reproducible quantitative analysis of RV volumes and function [8].
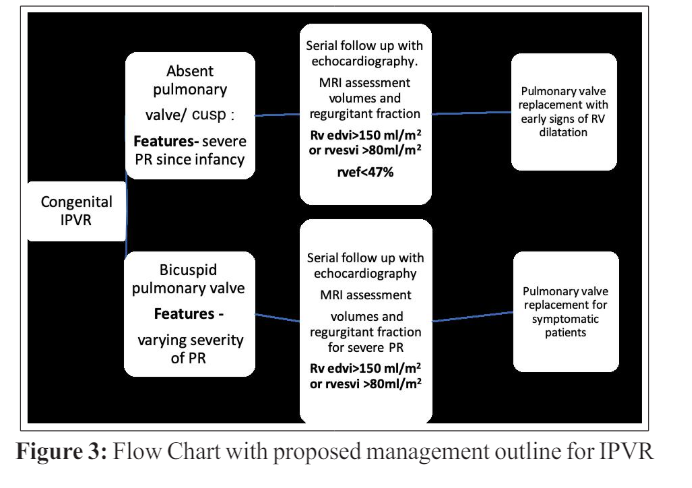
Extrapolating from the findings of the above-mentioned study, we propose that earlier intervention at RVEDI of >150 ml/m2 should offer a safer margin for RV normalization after PV replacement [8].
In an anatomy as in this case, when the valve is inherently regurgitant, the timing of PV replacement, should be with early signs of RV dilatation. Other considerations would be development of sustained atrial or ventricular arrhythmias, development of tricuspid regurgitation, or additional lesions needing bypass surgery. In late presenters, moderate to severe RV dysfunction is an indication for valve replacement. (Proposed management outline Figure 3).
Congenital IPVR due an absent single pulmonary cusp is a very rare entity. The timing of pulmonary valve replacement in this setting should be at the onset of RV dilatation to avoid progressive ventricular dysfunction.
Informed Consent: Obtained Institute`s ethical board issued a waiver for this case report.