Author(s): Anas Jawaid, Zaid Al-Jebaje* and Eduardo Arazoza
Abstract
Apical Hypertrophic Cardiomyopathy (ApHCM) is a variant of hypertrophic cardiomyopathy frequently found in Eastern Asian countries, but rarely seen in the United States. Patients typically present with symptoms of diastolic heart failure and characteristic electrocardiographic findings. Further risk-stratification is based on echocardiographic and cardiac MRI findings. Although classically thought to carry a favorable prognosis, recent evidence suggests a more nuanced approach to risk stratification and prognosis should be adopted. Here we present the case of a young male newly diagnosed with ApHCM and review the salient factors informing subsequent evaluation.
Apical Hypertrophic Cardiomyopathy (ApHCM) is a variant of hypertrophic cardiomyopathy frequently found in Eastern Asian countries, but rarely seen in the United States. Patients typically present with symptoms of diastolic heart failure and characteristic electrocardiographic findings. Further risk-stratification is based on echocardiographic and cardiac MRI findings. Although classically thought to carry a favorable prognosis, recent evidence suggests a more nuanced approach to risk stratification and prognosis should be adopted. Here we present the case of a young male newly diagnosed with ApHCM and review the salient factors informing subsequent evaluation.
A 39 year-old African American male with a past medical history notable for hypertension presented for outpatient cardiology consultation for atypical chest pain and an abnormal electrocardiogram.
The patient had initially presented to his primary care physician with a chief complaint of sudden-onset, intermittent pleuritic chest pain that was worse with exertion and improved with rest. He was noted to have a blood pressure of 147/90. An electrocardiogram obtained in the office revealed diffuse non-specific ST segment abnormalities that were unchanged from electrocardiograms obtained several years prior to presentation. The patient refused further evaluation in the Emergency Department. He consented to laboratory testing, which revealed a Troponin I of <0.01 and a BNP level of 8. A complete blood count, comprehensive metabolic panel, and a chest x-ray were without abnormalities. The patient was initiated on carvedilol 3.25 mg BID and referred for cardiology consultation.
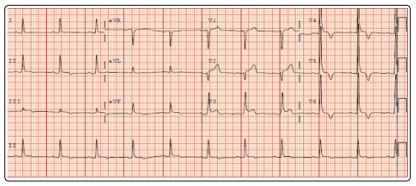
The patient presented to cardiology a week following his initial PCP visit. His blood pressure was 132/82. He reported that his chest pain had resolved without recurrence. The patient denied any family history of sudden cardiac death. An electrocardiogram was obtained, revealing left ventricular hypertrophy by the SokolowLyon criteria, as well as diffuse T-wave inversions (Figure 1).
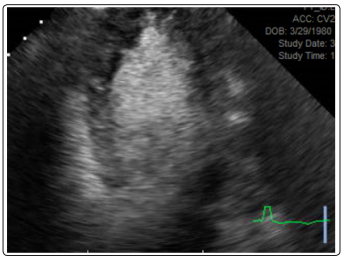
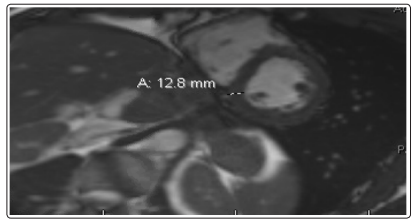
A stress echocardiogram was obtained, which was notable for absence of ischemia. However, the patient had the characteristic “Ace of Spades” appearance of the left ventricular cavity during diastole that is seen with apical hypertrophic cardiomyopathy (Figure 2). A cardiac MRI was subsequently obtained, which revealed circumferential thickening of the left ventricular apical segments, most prominent at the anterior, anteroseptal, septal, and inferoseptal walls (Figure 3). There was no abnormal late gadolinium enhancement. These findings were consistent with apical variant hypertrophic cardiomyopathy. He was scheduled for regular follow-up.
Apical hypertrophic cardiomyopathy (ApHCM) is a subset of the hypertrophic cardiomyopathy population, in which the hypertrophy is localized to the LV apex. Additional thickening of the midventricular and/or basal septum represent a mixed subtype of ApHCM. Each of these phenotypes can be associated with the presence of an apical aneurysm.
ApHCM was first described in Japan in 1976 as a novel condition characterized by giant negative T-waves, apical hypertrophy, and an ace-of-spades configuration of the LV cavity [1]. The prevalence of ApHCM is much higher in eastern Asian countries than in the United States; estimates have ApHCM representing up to 25% of all hypertrophic cardiomyopathy (HCM) patients in Japan, and 38% of all HCM patients in North Korea. In the United States, however, prevalence estimates place ApHCM between 3-11% of all HCM patients [2]. Typically, ApHCM is diagnosed in mid-life and a family history is present in about 6% of patients [3,4]. Like HCM, ApHCM involves mutations in genes coding for various sarcomeric and non-sarcomeric proteins although the final phenotypic presentation is likely multifactorial and involves modification of the genetic substrate [5]. Male gender appears to be affected preferentially in the Japanese population, but a gender predilection is not observed in other populations.
The cardinal clinical manifestations of ApHCM involve consequences of diastolic dysfunction. As such, patients may present with dyspnea on exertion or rest, exercise intolerance, or evidence of volume overload. Unlike classic HCM, there is rarely dynamic LV outflow tract obstruction or mitral regurgitation. However, ApHCM can lead to midventricular obstruction with cavity obstruction and apical aneurysm formation, which is independently associated with an increased risk of sudden cardiac death [6]. Myocardial ischemia can occur as septal branches of the left anterior descending artery are squeezed by the hypertrophic myocardium.
Diagnosis of ApHCM begins with finding typical EKG findings. In fact, ApHCM is the only specific hypertrophic pattern that had its own characteristic EKG findings with the giant T-wave inversions (>1 mV) and high voltages in the left precordial leads [7]. However, a recent case series revealed that only about 11%of patients feature these characteristic EKG findings [8]. The diagnostic criterion for the diagnosis of ApHCM is wall thickness greater than 15 mm of the LV apex or the ratio of apical to basal LV wall thickness of 1.3-1.5 [9]. The preferred initial test is a transthoracic echocardiogram (TTE) although image quality is dependent on multiple factors including sonographer expertise and patient body habitus. Contrast can be given to improve LV endocardial border definition if the LV is not well-visualized to obtain an “aces-of-spade” configuration of the apical segment. A complete echocardiographic assessment includes quantifying the presence, magnitude, and distribution of LV hypertrophy and identification of ApHCM variants. Echocardiography can also help identify mid-ventricular obstruction with cavity obliteration. If echocardiography produces suboptimal images, cardiac MRI can be used for assessment. An added benefit of cardiac MRI is the ability to identify apical aneurysms as well as replacement fibrosis through late gadolinium enhancement, which are both important prognostic factors. SPECT is another modality that may be utilized, with the characteristic “solar polar” pattern seen due to differences in uptake between the apex and surrounding myocardium in ApHCM [10].
Management of ApHCM is still evolving and hampered by the absence of adequately powered pharmacologic trials. As such, medical therapy is primarily focused on managing symptoms of heart failure. Beta blockers are usually the initial drug of choice given their ability to prevent arrythmias, decrease angina, and improve diastolic filling. Calcium-channel blockers can be attempted for similar reasons if beta-blockade is intolerable. Unlike ischemic cardiomyopathy, there is little evidence that blockade of the renin-angiotensin systems results in improved cardiac remodeling [11]. Interventional approaches, such as alcohol septal ablation, or transaortic septal myectomy, are precluded by ApHCM anatomy. However, novel surgical approaches involving apical myectomies may be used to improve functional status in select patients [12]. Additionally, anticoagulation may be considered in the presence of an apical aneurysm.
As in HCM as a broader entity, the biggest concern in ApHCM is that of sudden cardiac death. There are no specific guidelines for primary prevention of sudden cardiac death in the ApHCM. However, patients require careful evaluation for family history of sudden cardiac death, unexplained syncope during exercise, leftventricular wall thickness >3cm, and/or episodes of ventricular arrythmias. Additional stratification with the presence of late gadolinium enhancement on MRI and the presence of an apical aneurysm should be performed as both of these variables increase the risk of sudden cardiac death.
Although ApHCM had widely been considered to have a benign prognosis, more recent studies have suggested mortality rates higher than previously considered. A large cohort followed at Mayo Clinic for a mean of 6.5 years revealed an overall death rate of 29%, with significantly worse mortality in women compared to men (45% vs. 19%) [8]. Worse outcomes more frequently appear to be associated with the “mixed form” compared to the “pure apical” form, with patients more frequently presenting with higher LV filling pressures, higher BNP levels, and higher troponin levels in the absence of acute coronary syndromes [3,6]. As such, it is important to recognize the heterogeneity inherent even within cohorts of patients with ApHCM.
For our patient, initial echocardiographic evaluation was sufficient for a diagnosis of apical hypertrophy. A cardiac MRI was obtained to further evaluate the extent of apical thickening, and to determine the presence of fibrosis. Our patient’s CMR did not reveal abnormal late gadolinium enhancement, fibrosis, or the presence of an apical aneurysm. In the absence of high-risk features based on history and imaging findings, he was not offered an ICD. He remains in good health on a beta-blocker regimen with close follow-up.