Author(s): Mavra Fatima
Myelodysplastic syndrome (MDS) is a spectrum of bone marrow disorders resulting from clonal stem cell defect that manifest as cytopenias, ineffective hematopoiesis and dysplasia in all the three cell lines. Myelodysplastic syndrome is rare in children and accounts for less than 5% of the hematopoieitic malignancies below the age of 14 years. Refractory cytopenia of childhood (RCC) is the most common type of MDS that is seen in children. In this study, we report a case of 14 years old boy who presented to us with suspicion of aplastic anemia and was diagnosed as refractory cytopenia of childhood, a type of myelodysplastic syndrome.
The myelodysplastic syndromes are a diverse group of clonal stem cell disorders that are characterized by cytopenias, macrocytosis, morphological dysplastic features in bone marrow with an overall 40% risk of transformation into acute leukemia [1]. Median age for MDS is considered to be in the seventh decade and its occurrence in children is uncommon, accounting for less than 5% of all the hemopoietic malignancies in patients less than 14 years of age [2,3]. Primary MDS also called de novo MDS should be differentiated from secondary MDS that is associated with bone marrow failure syndromes (congenital or acquired) and cytotoxic therapy for any prior malignant or non malignant condition [3]. In the past MDS associated with Down syndrome comprised one fourth of the pediatric MDS [4]. The term “Refractory cytopenia of childhood” is given to MDS cases with persistent cytopenias, less than 5% of blast cells in bone marrow and less than 2% of blast cells in peripheral blood [5].
A 14 years old boy presented on 31st January, 2020 for bone marrow biopsy procedure at Chughtai Lab, Lahore with suspicion of aplastic anemia. Presenting complaints were: vomiting for the last 2 weeks, gum bleeding 4 months ago, low grade fever (off and on) for the last 2 yrs. He also complained of dyspnea on exertion, anorexia and undocumented weight loss. In the last 6 months, the patient had been transfused 4 units of whole blood and last transfusion was given 1 month prior to bone marrow biopsy procedure. He was clinically well and vitally stable. The only significant finding was remarkable pallor. No visceromegaly or lymphadenopathy was appreciated on examination. He had no rudimentary or absent thumbs, no significant x-ray findings or dysmorphic features.
Complete blood picture showed pancytopenia ( TLC: 3.57 x 109/L, Hb: 4.7 g/dl, Platelets: 28 x 109/L). Absolute neutrophil count was 1.2 x 109/L, reticulocyte count was 1% and absolute reticulocyte count was 75%.
RBCs showed dimorphic population with normochromic, normocytic and macrocytic population. WBCs showed Pseudo Pelger-Huet anomaly. Platelets were reduced on the smears, no platelet clumps were seen. However platelets showed normal morphology.
showed hypocellular bone marrow aspirate. Although overall cellularity of the marrow was reduced, a substantial number of hematopoietic cells were present on the smears. Erythroid precursors showed dysplastic features like megaloblastoid change, multinuclearity and karyorrhexis. Myeloid precursors also showed dysplastic changes like giant stab forms and metamyelocytes, Pseudo Pelger Huet anomaly and hypogranularity. Blast cells constituted about 2-3 % of nucleated marrow cells. Megakaryocytes were present and showed dysplasia (hypolobated megakaryocytes). Dysplasia was observed in all three cell lineages.
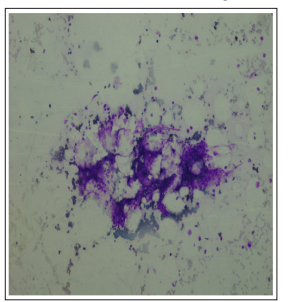
Figure 1: PMG Showing hypocellular BMA Fragment (20x)
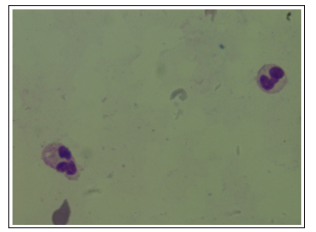
Figure 2: Pseudo Pelger Huet Anomaly in Neutrophils (100x)
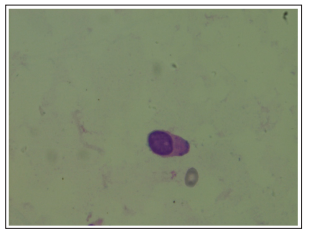
Figure 3: Blast Cell in Bone Marrow Aspirate (100x)
2 trephine biopsies were done from right and left posterior iliac crest which showed normal bony trabaculae and hypocellular bone marrow with patchy distribution i.e hypocellular areas alternating with a few cellular areas. Erythroid and myeloid precursors showed dysplastic changes. Megakaryocytes were prominent in the cellular areas and many micromegakaryocytes were also seen. No area of infiltration was seen at the site of biopsy.
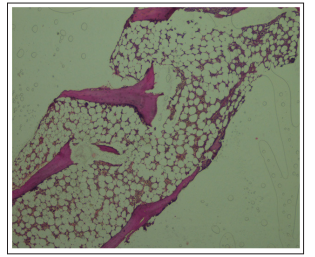
Figure 4: H&E Section of Trephine Biopsy Showing Patchy
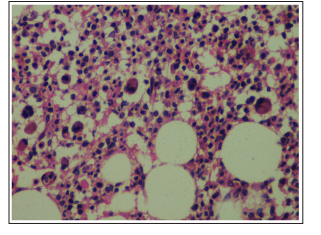
Figure 5: Trephine Biopsy Showing Hypolobated, Mononulcear Micromegakaryocytes
CD34: Positive in 3-4% of the nucleated marrow cells
CD61: Positive in micromegakaryocytes
Bone marrow findings were suggestive of refractory cytopenia of childhood (myelodysplastic syndrome) with multilineage dysplasia.
Karyotyping: Showed Monosomy 7 (Reported on Bone Marrow Aspirate.)
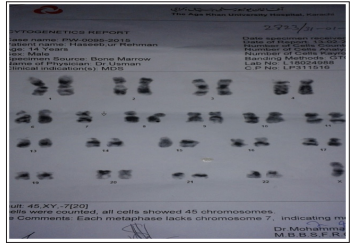
Chromosomal breakage studies and workup for Paroxysmal Nocturnal Hemoglobinuria (PNH) was not recommended due to morphological and cytogenetic evidence for RCC. The patient was counseled and referred regarding further treatment. Multiple unsuccessful attempts were made to contact the patient for follow up.
Childhood myelodysplastic syndrome is rare. RCC accounts for 50% of the cases of childhood MDS. Median age at presentation is 6.8 years [6]. Most appropriate treatment option is allogenic stem cell transplant and cure rate is around 60% [7].
It is difficult to differentiate between refractory cytopenia of childhood, bone marrow failure syndromes and severe aplastic anemia(SAA) as all of these disorders show hypocellular bone marrow [8]. Although a few cases show hypercellular bone marrow as well [5]. In our case, bone marrow aspirate was hypocellular and trephine biopsy showed patchy cellularity. On the basis of clinical features, morphology and cytogenetics, it is now possible to make an accurate diagnosis. In RCC, there are no skeletal abnormalities which are common in inherited bone marrow failure syndromes. Certain other features that are present in RCC and helped in the diagnosis of our case are the presence of patchy marrow cellularity, presence of erythroid islands along with dysplastic features like nuclear budding, karyorrhexis and internuclear bridging. Myelopoiesis show dysplasia like hyposegmentation with pseudo-Pelger-Huet nuclei and blast cells constitute <5% of the bone marrow cells which are not seen in aplastic anemia or any bone marrow failure syndromes [1,9]. If 5% or more CD34, myeloperoxidase, lysozyme and CD117 positive blast cells are seen it is suggestive of progression of disease to high grade MDS. Micromegakaryocytes are a strong indicator of RCC which are better seen by CD41 and CD61 that stain these small megakaryocytes [2,5]. All the above mentioned features are absent in bone marrow failure syndromes, hence we were able to make the diagnosis of RCC.
Karyotyping is a really helpful tool that suggests the progression of disease. Most common cytogenetic abnormality is monosomy 7 followed by trisomy 8 and trisomy 21 [4]. Presence of monosomy 7 suggests the probability of progression of disease whereas patients with trisomy 8 and normal karyotype may have a long stable course [7,10]. Presence of cytogentic abnormalities help to differentiate between refractory cytopenia, aplastic anemia and congenital or acquired bone marrow failure syndromes [9].