Author(s): Halo Yohans
This review provides an overview of immunodiagnostic techniques that are used for the diagnosis of microbial diseases. In particular it describes serological techniques by classifying it into two broad categories: Primary binding tests that directly measure the binding of antigen to antibody and secondary binding tests that measure the result of antigen-antibody interaction in vitro. Generally, results obtained from this review can increase the knowledge and skills of professionals for the diagnosis and detection of microbial diseases.
Immunodiagnostic techniques for detection of antigen or antibody immediately imply a variety of applications of immunological and immunochemical methods to the diagnosis of microbial diseases [1]. The immune responses of animals can be used in two general ways in the diagnostic laboratory. First, specific antibody may be used to detect or identify an antigen. This antigen can he associated with an infectious agent, or simply be a molecule that needs to be located or measured. Second, by detecting the presence of specific antibody in serum, it is possible to determine whether an animal has been previously exposed to a specific organism. This will assist in establishing a diagnosis or determining the degree of exposure of the population to that organism. The measurement of antigenantibody interactions for diagnosis purposes is called serology [2]. Serological techniques can be classified into two broad categories: Primary binding tests that directly measure the binding of antigen to antibody and secondary binding tests that measure the result of antigen-antibody interaction in vitro. These tests are usually less sensitive than the primary binding tests, but may be simpler to perform or require simpler technology. The most complex tests, in vivo tests, measure the actual protective effect of antibodies in an animal [3]. In addition to basic microbiological methods, such as microscopy and culture, to detect pathologic organisms, antigen or antibody detection methods by immunoassay are commonly used, and will be expanded further for rapid and accurate diagnosis of the common or newly emerging infection-causing agents in clinical as well as public health laboratories. Immunoassays have been developed with emphasis on fast and sensitive detection technologies and automated systems. Immunoassays for detection of host-produced antibodies directed against microorganisms, particularly viruses, are now one of the most widely used analytical techniques in laboratory medicine. [4]. Therefore, the objective of this review is discribing the principles and characteristics of major immunodiagnostic techniques and their application for the detection of microorganisms antige in the specimens.
The most common source of antibody for testing is serum obtained by allowing a blood sample to clot and the clot to retract. Serum may be stored frozen and tested when convenient. If necessary the serum can be depleted of complement activity, by heating to 56 0c for 30 minutes [3]. Complement is a normal constituent of all fresh serum, but the complement in fresh, unheated guinea pig serum is the most efficient in hemolytic tests. Serum used as a source of complement for serological applications should be stored frozen in small volumes. Once thawed, it should be used promptly. It should not be repeatedly frozen and thawed [3].
Because immunoglobulins are complex proteins they are antigenic when injected into an animal of a different species. For example, purified dog immunoglobulins can be injected into rabbits. The rabbits respond by making specific antibodies called antiglobulins. Depending on the purity of the injected immunoglobulin, it is possible to make nonspecific antiglobulins against immunoglobulins of all classes; or very specific antiglobulins directed against single classes. Antiglbulins are essential reagents in many immunological tests [5].
An antibody is a protein used by the immune system to identify and neutralize foreign objects like bacteria and viruses. Each antibody recognizes a specific antigen unique to its target. Polyclonal antibodies are antibodies that are derived from different cell lines. They differ in amino acid sequence and bind to multiple epitopes of all antigens used in the immunization. Monoclonal antibodies (mAb) are antibodies that are identical because they are produced by one type of immune cell, all clones of a single parent cell [6]. Monoclonal antibody production or mAb is produced by cell lines or clones obtained from the immunized animals with the substance to be studied. Cell lines are produced by fusing B cells from the immunized animal with myeloma cells. To produce the desired mAb, the cells must be grown in either of two ways: by injection into the peritoneal cavity of a suitably prepared mouse (the in vivo or mouse ascites method) or by in vitro tissue culture [12].
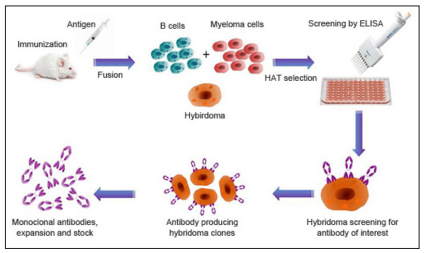
Figure 1: A Typical Monoclonal Antibody Production Process [12].
Hybridoma-derived monoclonal antibodies are pure and specific, can be used as standard chemical reagents to demonstrate the antigen of the causative agents or indirectly for serological detection of antibodies against the causative agents and can be obtained in almost unlimited amounts. As a result, monoclonal antibodies frequently replace conventional antiserum as reagents in immunodiagnostic tests [6].
Radioimmunoassay first introduced by Yalow and Berson in 1959 [7]. Radioimmunoassay is a quantitative test used for detecting specific antigens in patient serum. In this assay, the sample antigen is incubated with its complementary anti-bodies allowing them to bind [8]. Subsequently, the added radioactive labeled antigen competes with sample antigens (http://www.izotop.hu, 2017).
Competitive immunoassay are based on the principle that unlabeled antigen will displace radiolabeled antigen from immune complexes. These tests are sensitive and so; are commonly used to detect trace amount of drugs. The antigen or drug is labeled with an isotope such, as tritium (H3), carbon 14, or iodine 125 [3]. When radiolabeled antigen is mixed with its specific antibody, it combines to form immune complexes that may be precipitated out of solution. The radioactivity of the supernatant fluid is measure of the amount of unbound antigen. If unlabeled antigen is added to the mixture, it will compete with the labeled antigen for antibodybinding sites. As a result, some labeled antigen will be unable to bind, and the amount of radioactivity in the supernatant will increase. if a standard curve is first constructed based on the use of known amounts of unlabeled ‘antigen, then the amount of antigen in a test sample may be measured by reference to this standard curve of counts per minute bound per counts per minute free of the labeled antigen by a technique called extrapolation [8].
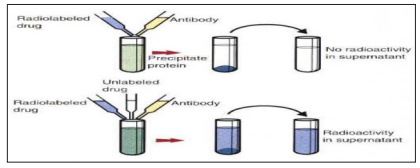
Figure 2: Principle of Radioimmunoassay Test [3].
Immunofluorescence (IF) is a histochemical laboratory staining technique that relies on antibodies–antigens interactions in tissue or body fluids. IF is broadly used in clinical immunology laboratories to help in the diagnosis of autoimmune diseases and in patients’ treatment and monitoring [9].
Fluorescent dyes are commonly employed as labels in primary binding tests, the most important being fluorescein isothiocyanate (FITC). FITC is a yellow compound that can be bound to antibodies without affecting their reactivity. when irradiated with invisible ultraviolet or blue light at: 290 and 145 µm, FITC reemits visible green light at 525 µm. FITC-labeled antibodies are used in the direct and indirect fluorescent Antibody tests. The IF test is available in four basic types, direct IF, indirect IF (the most commonly used in the clinical immunology laboratory), indirect IF complement fixation, and double IF [3].
Direct IF is a technique where fluorescently labeled antibody specific to the target antigen is used in a patient tissue or cell. It is a one-step procedure that uses only one primary conjugated antibody. Antibody directed against a specific antigen such as a Bacterium or virus is first labeled with FlTC. A tissue or smear containing the organism is fixed to a glass slide incubated with a labeled antiserum, and washed to remove unbound antibody [3]. When examined by dark field illumination under a micro-scope with an ultraviolet light source, the organisms that bind the labeled antibody fluoresce brightly. This test can identify bacteria when their numbers are every low [11].
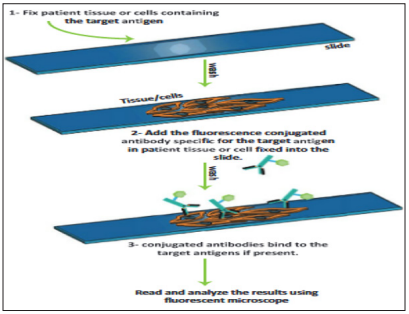
Figure 3: Principle of Direct Immunofluorescence (If) [11].
Indirect IF is usually used to detect specific patient antibodies. It is performed in two steps, the first step using unlabeled antibodies (patient antibodies), and the second step requires using secondary antibodies that are specific to the Fc region and labeled with fluorescence [9]. Indirect fluorescent antibody tests are used to detect antibodies in serum or to identify antigens in tissues or sell cultures. When testing for antibodies, antigen is employed as a tissue smear, section, or cell culture on a slide or cover slip. This is incubated in a serum suspected of containing antibodies to that antigen. The serum is then washed off, leaving only specific antibodies bound to the antigen. These bound antibodies may be visualized after incubating the smear in FITC-labeled antiglobulin. When unbound antiglobulin is removed by washing and the slide examined, f1uorescence indicates that antibody was present in the test serum. The quantity of antibody in the test serum may be estimated by examining increasing dilutions of serum on different antigen preparations [3].
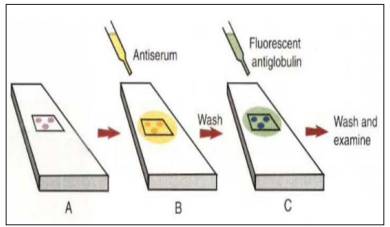
Figure 4: Principle of Indirect Immunofluorescence (IFI) [9].
Indirect immunofluorescence complement fixation (IF-CF) is more sensitive than indirect IF because it uses an amplification principle. Here, the generation of antigen–antibody complexes activates the complement system to release C3 molecules that are detected by anti-C3 conjugated to fluorochrome [13].
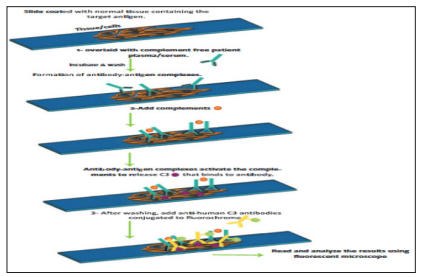
Figure 5: Principle of Indirect If Complement Fixation [13].
Double immunofluorescence (DIF) allows the detection of two different antibodies on cells by using two specific antibodies for the target antigens. Each is labeled with a different fluorochrome such as FITC (fluorescein) and rhodamine. This technique can be used with direct and indirect IF methods [9].
Among the most important immunoassays employed in veterinary medicine are Enzyme linked immunosorbent immunoassays. ELISA was invented by Peter Perlmann and Eva Engvall at Stockholm University in Sweden. It is a plate-based assay technique designed for detecting and quantifying peptides, proteins, antibodies and hormones. ELISA utilizes enzyme-labeled antigens and antibodies to detect the biological molecules, the most commonly used enzymes being alkaline phosphatase and glucose oxidase [14].The antigen in fluid phase is immobilized, usually in 96-well microtiter plates. The antigen is allowed to bind to a specific antibody, which is itself subsequently detected by a secondary, enzyme-coupled antibody. A chromogenic substrate for the enzyme yields a visible color change or fluorescence, indicating the presence of antigen. ELISAs can be performed with a number of modifications to the basic procedure: direct, indirect, sandwich or competitive [15]. A direct ELISA assay would detect the amount of antigen on a multi-well plate using enzyme conjugated antibodies, and is rarely used. This version of ELISA is the method of choice to detect the presence of serum antibodies against human immunodeficiency virus and a good option if there is no commercially available ELISA kit for your target protein [4].
For indirect detection, the antigen coated to a multi-well plate is detected in two stages or layers. First an unlabeled primary antibody, which is specific for the antigen, is applied. The primary antibody is not conjugated, then indirect ELISA is required in which a conjugated secondary antibody is targeted to the isotope of the primary antibody. Next, an enzyme-labeled secondary antibody is bound to the first antibody. The secondary antibody is usually an anti-species antibody and is often polyclonal [16]. This system has been widely applied in diagnostics because it allows large number of samples to be screened with a single conjugated secondary antibody. A main disadvantage of indirect ELISA is that the method of antigen immobilization is not specific. When serum is used as the test antigen, all proteins in the sample may adhere to the wells of a microtiter plate [17].
The sandwich technique is used to identify a specific sample antigen. Sandwich ELISAs typically require the use of matched antibody pairs, where each antibody is specific for a different, nonoverlapping part (epitope) of the antigen molecule. A first antibody (capture antibody) is coated to the wells. The sample solution is then added to the well. A second antibody (detection antibody) follows this step in order to measure the concentration of the sample (https:// www.bosterbio.com/ebooks). In this test, the intensity of the color reaction is related directly to the amount of bound antigen. Because these tests involve the formation of antibody-antigen-antibody layers, they are called sandwich ELlSAs.
Sandwich ELISAs are used to detect circulating virus in blood from cats with feline leukemia [14]. A competitive ELISA can be used to measure hapten molecules or viral antigens. In this technique, antibody coats the inside wall of the micro well. In a single reaction, antigen from the test sample and enzyme-labeled antigen compete for the antibody binding sites. The amount of labeled antigen bound to the solid phase (microwell) is inversely related to the concentration of antigen present in the test sample. This technique clearly has some speed advantages over other ELISA techniques. It can be made very sensitive if the sample antigen is permitted to react with the antibody before the labeled antigen conjugate is added [15].
Immunofiltration and immuno chromatography are most common recent Developments in Antibody-Antigen Immune Testing. Simple immunoassays that give results in minutes and are useful in determining a preliminary diagnosis. Immunofiltration is rapid ELISA that uses antibodies bound to membrane filters rather than polystyrene plates. Membrane filters have large surface area. Immuno chromatography is very rapid and easy-to-read ELISAs. Antigen solution flows through a porous strip and encounters labeled antibody. Visible line produced when antigen-antibody immune complexes encounter antibody against them. Used for pregnancy testing and rapid identification of infectious agents [3].

Figure 6: Principle of Indirect, Sandwich and Competitive Elisa [17].
The detection principle of chemiluminescence immunoassays (CLIA or LIA) is similar to that of ELISA. The antibodies or antigens are detected by measuring the specific wavelength of light produced through a chemical reaction. Chemiluminescent substrates include acridin ester or isoluminol which oxidize in the presence of hydrogen peroxide and a catalyzer to produce the luminescence signal. [18].
The electro-chemiluminescence immunoassay (ECLIA) is a further development of CLIA. As with CLIA, ECLIA uses a chemiluminescence reaction to detect the antibodies or antigens being tested for, however the chemiluminescent substrate (luminophore) oxidizes by applying an electrical current rather than as a result of a chemical reaction. The detection sensitivity of the CLIA and ECLIA methods is higher than for ELISA. Additional advantages of the CLIA and ECLIA methods include a shorter analysis time than with ELISA, and automatable processing. CLIAs and ECLIAs are currently used in fully automated analysis systems produced by various manufacturers. [20].
Immunoblotting which includes Western blot is another technique for antibody detection. Western blotting identifies and provides preliminary quantitation of a specific protein in a complex mixture of proteins. Capture antigens such as proteins are electrotransferred to a nitrocellulose membrane. If target antibodies are present in the specimen, they will bind to the antigens present on the nitrocellulose strips. Visualization of the antibodies bound to antigen is accomplished using a series of reactions with goat antihuman IgG conjugated with biotin, avidin conjugated with HRP, and the HRP substrate. The bands corresponding to the antigens will be seen on the nitrocellulose strip [21]. Western blotting has proven very useful in identifying the important antigens in complex microorganisms or parasites [3].
A variation of the western blot is the dot blot. It represents a simplification of the western blot method, with the exception that the proteins to be detected are not first separated by electrophoresis. Dot blot utilizes a dry nitrocellulose or PVDF membrane that has been “dotted” with sample homogenate. The presence of the antigen can be determined using specific antiserum and enzymelabeled antiglobulin in sequence. After exposure to enzyme substrate, the presence of a stained dot is a positive reaction. Use of nasal washings as a source of the antigen, such as when detecting respiratory viruses, is called a snot-blot. This particular technique is usually used for detection and quantification. The technique also provides a quick way to determine if an antibody is non-specific, particularly for a secondary antibody. However, it offers no information on the size of the target protein [22].
Blot methods offer the advantage of being able to specifically detect a number of antigen-specific antibodies in one test assay. Native and recombinant antigens can be jointly used on the same blot strip. By using defined antigens, blot methods exhibit a high specificity when it comes to detecting antibodies. Therefore, blot methods are more often used as confirmatory tests in the stepwise approach to diagnostic testing (e.g. borreliosis diagnosis) or as additional tests (e.g. toxoplasmosis, CMV, EBV diagnosis) [18].
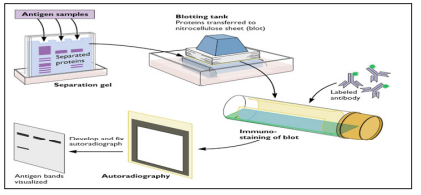
Figure 7: Western Blotting Techniques [22].
Immunohistochemistry employs antigen: antibody reactions and is used primarily by pathologists and researchers. Antibodies specific to a target of interest in the tissue are applied and detected, often using an enzymatic reaction. Using a range of enzymes with different substrates can allow the deposition of products of different colors that reflect the binding of different antibodies. Various chemical additives are available that can enhance the density and/or the color of the staining. This allows pathologists to recognize certain markers in tissues, such as cytokeratin and vimentin, which identify cells of epithelial vs mesenchymal origin, respectively, and use immunohistochemistry to identify whether lymphoma is of B- or T-cell origin from a lymph node or other tissue biopsy [17]. Many other specialized markers identifying specific cellular antigens can reveal further information about the tissue samples; these are the “special stains” that allow a more detailed evaluation of histopathologic biopsies [16].
In immunohistochemistry, the samples are prepared by sectioning intact tissue and the stained cells are therefore localized in their biological context. Because the sample is prepared by sectioning (cutting), immunohistochemical samples need not be permeabilized by detergent treatment prior to staining, as the cut surfaces of cells allow antibody to reach intracellular targets [17].
The reactions between antigens and antibodies are commonly followed by a secondary reaction. Thus if antibodies combine with soluble antigens in solution, the resulting complexes may precipitate. If antigens are particulate (e.g.bacteria or red blood cells), then antibodies may make them clump or agglutinate. If an antibody can activate the classical complement pathway and the antigen is on a cell surface, then cell lysis may result [3].
Immunoprecipitation (IP) is a precipitation technique used to purify and enrich the protein of interest out of a protein mixture. IP isolated proteins can then further be analysed by Western blotting, ELISA, and mass spectrometry. IP helps to identify: presence, up/down regulation, size, stability, and interactions of the protein-of-interest [4].
The immunoprecipitation technique detects soluble antigens that react with antibodies called precipitins. The precipitin reaction occurs when bivalent or multivalent antibodies and antigens are mixed in the proper proportions. The antibodies link the antigen to form a large antibody-antigen network or lattice that settles out of solution when it becomes sufficiently large. Immunoprecipitation reactions occur only at the equivalence zone when there is an optimal ratio of antigen to antibody so that an insoluble lattice forms. If the precipitin reaction takes place in a test tube, a precipitation ring forms in the area in which the optimal ratio or equivalence zone develops [17].

Figure 8: Schematic for Immunoprecipitation (Ip) protocol (Source: mbl medical & biological lab co., ltd, 2017)
Immunodiffusion is a technique for the detection or measurement of antibodies and antigens by their precipitation which involves diffusion through a substance such as agar or gel agarose. Simply, it denotes precipitation in gel [3].
Based on the method employed, immune diffusion classified as radial immunodiffusion and ouchterlony double diffusion. Radial immunodiffusion (RID) or mancini method is a single diffusion technique whereby a solution containing the antigen is placed into wells in a gel or agar surface evenly impregnated with antibody. The diameter of the ring that precipitates around the well as a result of antigen antibody reaction corresponds to the amount of antigen in the solution.
Double diffusion in 2 dimensions according to Ouchterlony has been commercially available and a proven test for decades. Antigens and antibodies are pipetted into pre-punched wells on slides coated in agar. The principle behind the test is that compatible reaction partners diffuse to one another and precipitates become visible. The precipitates can be made more visible using an Amido black staining solution [21].
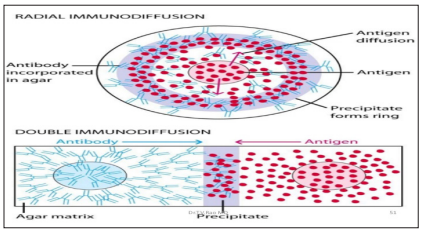
Figure 9: Diagrammatic Representation of Radial Immunodiffusion (Mancini Method) and Double Immundiffusion (Ouchterlony Method) In a Gel. (Source: kuby immunology, 2013)
The advantage of this method is that no equipment is needed except for the reference antigens and antibodies, the punch-cut agar, a humidity chamber and, possibly, an incubator. The disadvantage is that precipitating antibodies can only be detected if there is an equivalent antigen to antibody ratio, and readings are subject to individual, visual fluctuations. ID is primarily used to diagnose fungal infections caused by the pathogens of imported systemic mycoses and the pathogens of aspergilloma [17].
Some antigen mixtures are too complex to be resolved by simple diffusion and precipitation. Greater resolution is obtained by the technique of classical immunoelectrophoresis in which antigens are first separated based on their electrical charge, then visualized by the precipitation reaction. In this procedure antigens are separated by electrophoresis in an agar gel. Positively charged proteins move to the negative electrode, and negatively charged proteins move to the positive electrode. A trough is then cut next to the wells and filled with antibody. The plate is incubated; the antibodies and antigens will diffuse and form precipitation bands or arcs that can be better visualized by staining. This assay is used to separate the major blood proteins in serum for certain diagnostic tests [16].
Numerous procedures have been developed to detect antigen by means of the agglutination (clumping) of an artificial carrier particle or insoluble matrix, such as a latex bead, with antibody bound to the surface. These assays are classified as indirect agglutination reactions, also referred to as reverse passive agglutination. In addition, agglutination assays that detect an intact antigen directly on an organism’s surface or cell are classified as direct agglutination assays [23]. The assays use an inactivated whole organism mixed with patient serum to identify antibodies that indicate exposure to the infectious agent. Specific antibodies bind to surface antigens of the bacteria in a thick suspension and cause the bacteria to clump in visible aggregates. Such antibodies are called agglutinins, and the test is referred to as bacterial agglutination [24].
Electrostatic and additional chemical interactions influence the formation of aggregates in solutions. Because most bacterial surfaces have a negative charge, they tend to repel each other. Performance of agglutination tests in sterile physiologic saline (0.9% sodium chloride in distilled water), which contains free positive ions, enhances the ability of antibody to cause aggregation of bacteria. Although bacterial agglutination tests can be performedon the surface of plastic-coated reaction cards and in test tubes, tube agglutination tests, despite being more sensitive because a longer incubation period can be used, allowing more antigen and antibody to interact, are rarely used in the modern clinical laboratory [25].
Agglutination methods utilize the antibody-antigen bond to create clumping (agglutination) of particles. Agglutination tests to detect antigens employ fixed red cells (hemagglutination), latex beads, gelatin, or synthetic microbeads coated with specific antibody as carrier or indicator particles. In a typical agglutination assay for detection of microbial antigen, a drop of liquid suspension of antibody-coated particles is placed on a card, and the specimen is added and mixed. The card is then incubated, often on an oscillating mixer, and read by visually observing the clumping reaction. No washing is required. Agglutination assays can be made semiquantitative by performing serial dilutions of the specimen and reporting the greatest dilution which results in a positive reaction [2].
When particulate antigens combine with its homologous antibody, in the presence of an electrolyte at a suitable temperature and pH, the particles are clumped or agglutinated. Agglutination reactions are very sensitive, relatively easy to read and available in great variety. Zone phenomenon may be seen either in antigen excess or antibody excess. Agglutination occurs optimally, when antigens and antibodies react in equivalent proportion [10].
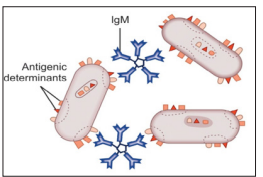
Figure 10: An Agglutination Reaction. When Antibodies React With Antigenic Determinant Sites on Antigens Carried on Neighboring Cells, Such as These Bacteria (or Red Blood Cells).
The particulate antigens (cells) agglutinate. IgM, the most efficient immunoglobulin for agglutination is shown here, but IgG also participates in agglutination reactions. (Source: Textbook of immunology, 2014) A major source of error in agglutination tests is the prozone reaction, which occurs when antigen is present in excess. “Prozoning” is observed at high antigen concentrations where excess antigen occupies most antibody-binding sites with unique antigen molecules, thus preventing the multiple antibody binding of each antigen that causes the particles to clump. These false-negative reactions can be detected by repeating the test at a higher dilution of sample, which reduces the antigen concentration into the range that produces agglutination [4].
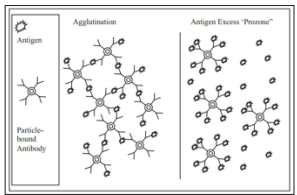
Figure 11: Particle Agglutination and Prozone Effect (Source: Advanced Techniques in Diagnostic Microbiology, 2018)
Direct agglutination of whole pathogens can be used to detect antibodies directed against the pathogens. The most basic tests measure the antibody produced by the host to antigen determinants on the surface of a bacterial agent in response to infection with that bacterium. In a thick suspension of the bacteria, the binding of specific antibodies to surface antigens of the bacteria causes the bacteria to clump together in visible aggregates. This type of agglutination is called bacterial agglutination [2]. The formation of aggregates in solution is influenced by electrostatic and other forces; therefore, certain conditions are usually necessary for satisfactory results. The use of sterile physiologic saline with free positive ions in the agglutination procedure enhances the aggregation of bacteria because most bacterial surfaces exhibit a negative charge that causes them to repel each other. Because it allows more time for the antigen-antibody reaction, tube testing is considered more sensitive than slide testing. The small volume of liquid used in slide testing requires rapid reading before the liquid evaporates [28].
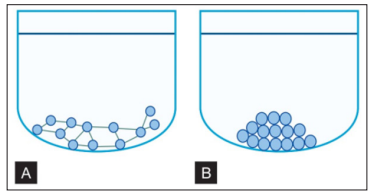
Figure 12: A And B: Direct Agglutination Test Showing Positive and Negative Reaction
a. In a positive (agglutination) reaction, sufficient antibodies are present in the serum to link the antigens together forming a mat of antigen-antibody complexes on the bottom of the well; B. In a negative (no agglutination) reaction, not enough antibodies are present to cause the linking of antigens. The particulate antigens roll down the sloping sides of the well, forming a pellet at the bottom. (Source: Textbook of immunology, 2014).
In Bacterial agglutination: Antiserum is added on a uniform suspension of pure bacteria. If there is clumping as well as clearing of the suspension, then the agglutination is positive. Control should be done along with the test with normal saline. Slide agglutination is routine procedure for the identification of bacterial isolates from clinical specimen. The other method of slide agglutination is red cell agglutination. It is also the method used for blood grouping and cross matching. Widal’s slide agglutination test: it is used for the diagnosis of enteric fever by Salmonella (S. typhi, S. paratyphi A and S. paratyphi B). Tube agglutination This is a standard quantitative method for the measurement of antibodies and used to Measure the Level of Antibodies to Particulate Antigens [10].
Serial dilutions are made of a sample to be tested for antibody. Then a fixed number of RBC’s/Bacteria/other such Particulate Antigen is added. Then the maximum dilution that gives agglutination is determined. Tube agglutination is routinely employed for the diagnosis of enteric fever, brucellosis, typhus fever, infectious mononucleosis and many other conditions. The tube agglutination test for brucellosis may be complicated by the prozone phenomenon and the presence of blocking antibodies. Several dilutions of the serum should be tested to prevent false negative reaction. Incomplete or blocking antibodies may be detected by antiglobulin (Coombs’) test [28].
Antibodies against soluble antigens can be detected by agglutination tests, if the antigens are adsorbed onto particles such as bentonite or latex particles. Such tests known as latex agglutination tests are commonly used for the rapid detection of the serum antibodies against bacterial and viral diseases. In such indirect (passive) agglutination tests, the antibody reacts with soluble antigen adhering to the particles. The particles then agglutinate with one another. The same principle can be applied in reverse by using particles coated with antibodies, to detect the antigens against which they are specific (e.g. streptococci in sore throat). Latex agglutination tests both passive and reverse passive are widely employed in the clinical laboratory (HBsAg, ASO, CRP, RA factor, classic pregnancy test, etc.) [3]
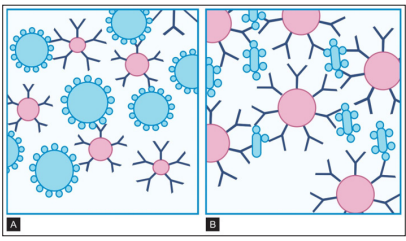
Figure 13: A and B: Reaction in Indirect Agglutination Tests
These tests are performed using antigens or antibodies coated onto particles such as minute latex spheres. A. When particles are coated with antigens, agglutination indicates the presence of antibodies such as the IgM shown here; B. When particles are coated with monoclonal antibodies, agglutination indicates the presence of antigens (Source: Textbook of immunology, 2014).
In latex agglutination procedures, antibody molecules can be bound to the surface of latex beads.Many antibody molecules can be bound to each latex particle, increasing the potential number of exposed antigen-binding sites. If an antigen is present in a test specimen such as C-reactive protein, the antigen will bind to the combining sites of the antibody exposed on the surface of the latex beads, forming visible cross-linked aggregates of latex beads and antigen. In some procedures (e.g., pregnancy tests, rubella antibody testing), latex particles can be coated with antigen. In the presence of serum antibodies, these particles agglutinate into large visible clumps [29].
Procedures based on latex agglutination must be performed under standardized conditions.Te amount of antigen-antibody binding is influenced by factors such as pH, osmolarity, and ionic concentration of the solution. A variety of conditions can produce false-positive or false-negative reactions in agglutination testing. Coagglutination and liposome-enhanced testing are variations of latex agglutination Co agglutination use antibodies bound to a particle to enhance the visibility of agglutination. It is a highly specific method but may not be as sensitive as latex agglutination for detecting small quantities of antigen [2].
The hemagglutination method of testing detects antibodies to erythrocyte antigens. The antibody-containing specimen can be serially diluted and a suspension of red blood cells (RBCs) added to the dilutions. If a suffcient concentration of antibody is present, the erythrocytes are cross-linked and agglutinated. If non-reacting antibody or an insufficiency quantity of antibody is present, the erythrocytes will fail to agglutinate [29]. By binding different antigens to the RBC surface in indirect hemagglutination or passive hemagglutination (PHA), the hemagglutination technique can be extended to detect antibodies to antigens other than those present on the cells. Chemicals such as chromic chloride, tannic acid, and glutaraldehyde can be used to cross-link antigens to the cells. Some antibodies (e.g., immunoglobulin G [IgG]) do not directly agglutinate erythrocytes. Tis incomplete or blocking type of antibody may be detected by using an enhancement medium such as antihuman globulin (AHG) reagent (also known as Coombs reagent).If AHG reagent is added, this second antibody binds to the antibody present on the erythrocytes [26].
Hemagglutination, is the clumping of red blood cells, by either a direct or indirect mechanism. This type of agglutination reaction is used in immunohematology for blood group typing (direct) or the detection of a red cell antibody (indirect). Hemagglutination is also commonly used in virology. The monospot test is a hemagglutination assay that detects heterophile (nonspecifc antibodies) produced in the early stages of infection with EpsteinBarr virus. More recently, indirect hemagglutination assays that use antigen from the infectious agent attached to a latex bead have been used to detect antibodies to human immunodefciency virus (HIV), T. pallidum, and hepatitis viruses (A, B, and C). Certain viruses such as those causing mumps, measles and influenza, have the ability to agglutinate RBCs without an antigen-antibody reaction; this process is called “viral hem-agglutination”. This type of hemagglutination can be used to detect antibodies that neutralize the agglutinating viruses in patient’s sera [24].
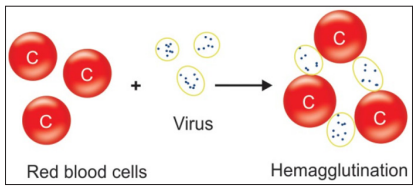
Figure 14: viral hemagglutination principle. (source: textbook of immunology, 2014)
The complement fixation test is a blood test in which a sample of serum is exposed to a particular antigen and complement in order to determine whether or not antibodies to that particular antigen are present. The nature of complement is to react in combination with antigen–antibody complexes. The relative lack of antigen specificity allows complement to react with almost any antigen– antibody complex. In the test procedure, complement remains free unless fixed by the particular antigen and antibody system in question. The indicator used in many complement fixation assays is sheep RBCs. In a positive or reactive test, the complement is bound to an antigen–antibody complex and is not free to interact with target RBCs. The RBCs remain unlysed and settle to the bottom of the well to form a button. In a negative or nonreactive test, the complement remains free to interact with the blood cells causing them to lyse [30].
The test is performed in two parts. First, antigen and antibodies (the serum under test deprived of its complement by heating at56 ?C) are incubated in the presence of normal Guinea pig serum as a source of complement. (Guinea pig serum is most commonly used because its complement lyses sheep red cells well.) After the antigen- antibody- complement mixture reacts, the amount of free complement remaining in the mixture is measured by adding an indicator system consisting of antibody-coated sheep red cells. Lysis of these cells (seen as the development of a transparent red solution) is a negative result because it indicates that complement was not activated and that antibody was absent from the serum under test [3]. The complement test is a powerful tool to identify antibodies reacting with antigens, and it permits confirmation of exposure to a specific microorganism [30].
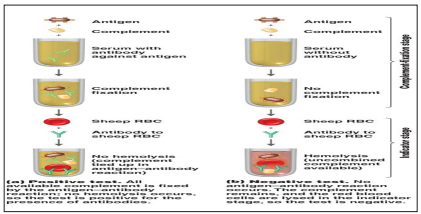
Figure 15: Complement Fixation Assay Principle
Complement may cause membrane damage, not only to erythrocytes but also to nucleated cells and to protozoa. Antibodies against cell surface, antigens thus may be measured by reacting target cells with antibody and complement, and estimating the resulting cell death. This form of assay is employed in the identification of major histocompatibility complex class I molecules [3]. Cytotoxicity tests are one of the most important indicators of an in vitro biological evaluation system. Furthermore, because of advances in cell biology, experimental methods to assess cytotoxicity are being continuously developed [31].
Neutralization tests are performed in vitro, at the cellular level including on cells in the form of organs and at the sub-cellular level. They assess the efficacy of antibody fragments, or antibodies, to inhibit critical stages of activities. Neutralization tests mostly apply to viruses and toxins Antibodies may effect virus neutralization by several mechanisms. They may prevent virus adsorption to cell receptors. A more frequently used neutralization test is the viral hemagglutination inhibition (HI) test. This test is used in the diagnosis of influenza, measles, mumps and a number of other infections caused by viruses that can agglutinate RBCs. If a patient’s serum contains antibodies against these viruses, the antibodies will react with viruses and neutralize them [10]
Bacterial exotoxins are good antigens and induce antibody (antitoxin) formation. Toxin neutralization may occur in vivo (Schick test) and is based on the ability of the circulating antitoxin to neutralize the bacterial toxins injected intradermally. This test indicates susceptibility or immunity. An example of toxin neutralization in vitro is the antistreptolysin O titer test. In this test, the antitoxin present in patient’s sera neutralizes the hemolytic activity of the streptococcal O hemolysin.
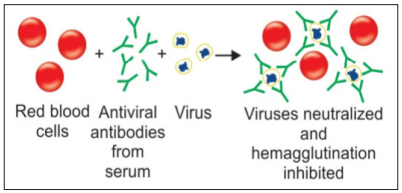
Figure 16: Reactions in Neutralization Tests [10]
Obviously, the presence of antibodies to a specific organism in animal’s serum indicates previous exposure to an epitope present on that organism. It does not, however, prove that infection exists or that any concurrent disease is actually caused by the organism in question. An immune response to an invading microorganism generates specific antibodies. These are a heterogeneous mixture. The specificity of the serum will reflect the dominant specificity of the antibody mixture. For example, the fact that the sera of most healthy horses contain antibodies to salmonella entericavar: typhimrium does not prove that most horses have salmonellosis. Thus, the presence of antibodies to an organism in a single serum sample is rarely of diagnostic significance. Only if at least two samples are taken 1 to 3 weeks apart and at least two a fourfold rise in titer is shown can a diagnosis be made. This should be done only in conjunction with careful analysis of clinical factors.
A second feature that must be considered in the interpretation of serological results is the possibility of errors. Technical errors are usually prevented by incorporation of appropriate controls into the test system. Other errors, however, are largely unavoidable. These may be of two types: false-positive results and false-negative results. a test in which a large proportion of the positive results is false considered to be nonspecific, whereas one with a very high proportion of false-negative results is considered to be insensitive. In general, the level of such errors is set by the criteria used to differentiate positive from negative reactions.
If these criteria are adjusted so that the number of false-positive results is reduced, then there will be an increasing proportion of false-negative results encountered, and vice versa. Thus, highly sensitive rests tend to be relatively nonspecific and highly specific tests are generally insensitive. The establishment of criteria in reading tests and from this, the sensitivity and specificity of a test are determined both by the requirements of the test procedure and by the importance of false-positive and false-negative reactions. In ideal tests it would be desirable for the criteria used in interpreting the test results to be so obvious and absolute that each rest would be absolutely sensitive and specific. Unfortunately, such ideal tests are uncommon.
The selection of a diagnostic test represents a compromise among its sensitivity, its specificity, and its complexity, that is, the number of steps involved, the degree of technical expertise required, its cost, and the nature of the equipment needed to conduct the test. Although precise guide lines cannot be drawn, it is usually most appropriate to use the most sensitive and specific test that can be satisfactorily performed with the available technical assistance and equipment, at the lowest cost [3].