Author(s): Elharrouni Alaoui Aicha*, Baybay Hanane, Douhi Zakia, Elloudi Sara and Mernissi FZ
IFAP syndromeis a rare autosomal recessive X-linked disease characterized by the triad of alopecia universalis, severe photophobia, and follicular ichthyosis. It is caused by loss of function of the gene MBTPS2. Its severity varies and there are only a few reports in the literature. We present a patient with characteristic clinical features.
IFAP syndrome is a rare genetic disorder characterized by ichthyosis and alopecia from birth and sometimes accompanied by short stature, intellectual disability, and seizures epilepsy that develop in the first few years of life. Photophobia may also be present in the first year of life or appears in infancy or early childhood. Its mode of inheritance is X-linked recessive, thus mostly affecting males. Affected or carrier females may display some of its clinical features.
An 18-month-old boy, has non consanguineous parents. Pregnancy was uneventful and the boy was born underweight and preterm at 36 weeks and 5 days by natural birth and there was no history of similar illness in the family or familiar history of atopy. He was the first birth in his family. He was referred to the dermatology department with congenital universal alopecia. The clinical examination revealed a photophobia, prominent forehead, large ears with staturo-ponderal delay and in the dermatological examination showed skin phototype II, spiky follicular ichthyosis on the scalp and extensor aspects of the extremities, including elbows and knees, generalized dryness of the skin, angular cheilitis, follicular papules and pustules on an erythematous base scattered, plantar keratoderma, associated with universal alopecia of scalp hair, eyebrows, and eyelashes, periungueal inflammation, dystrophic nail (Figure 1).
Histopathological examination of the cutaneous biopsy showed dilated hair follicles with keratin plugs extending above the surface of the skin, decreased or absent sebaceous glands and normal sweat glands, with the presence of the rare miniaturized atrophic hair follicles including rare dystrophic hair rods, giving the appearance of follicular keratosis with non-cicatricial alopecia in the context of the IFAP syndrome. He was observed by a Medical Genetics consultant where the IFAP syndrome was retained and he underwent conservative treatment with moisturizers and topical steroids and eye lubrication, with a good improvement.
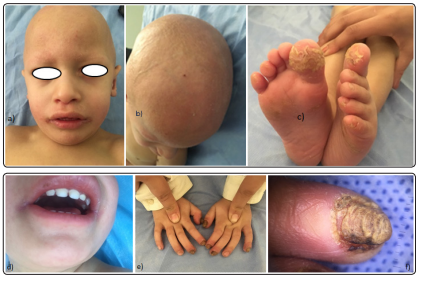
Figure 1: Clinical appearance and dermatologic findings of the patient. Note the alopecia with an absence of scalp hair, eyebrows, and lashes (a),(b). Spiky follicular hyperkeratosis on the trunk. Skin was dry and scaly (a),angular cheilitis (b), plantar keratoderma (c) and dystrophic nails (e),(f).
Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome (OMIM 308205) is an extremely rare X-linked oculocutaneous genetic disorder characterized by the triad of hyperkeratotic follicular papules, total to subtotal alopecia, and photophobia of varying degrees [1]. This association was first described as a syndrome by MacLeod in 1909 [1]. Since; only 37 cases of IFAP syndrome have been reported in the literature [2-4]. And most of these had additional features including atopic eczemain 36% cases [5].
In clinical presentation, Follicular ichthyosis predominantly distributed on the scalp and extensor aspects of the extremities, including elbows and knees, represents the classical cutaneous manifestations, which could give the skin “nutmeg grater-” or “sandpaper-” like texture [6]. Congenital alopecia involving the scalp, eyebrows and eyelashes is another essential cutaneous manifestation of IFAP.A non-cicatricle complete body alopecia is another characteristic dermatological finding of this syndrome [6]. Variable degrees of a collodion membrane may be present in the neonate. Psoriasiform plaques, angular cheilitis, periungueal inflammation, dystrophic nails, hypohidrosis, and atopic eczema was also described [6]. The palms and soles can by affected, as seen in our case [7]. Photophobia is an essential feature for the diagnosis of IFAP. It can be present early in life or later in childhood, but other Ophthalmological manifestations have been reported as well; corneal erosions, scarring atopic keratoconjunctivitis, nystagmus, and myopia [2]. Growth retardation, represented by short statue, is often observed, as seen in our case [8,9]. Psychomotor developmental delay, seizures and hypotonia were also noted [10,11]. Nevertheless, our patient did not show neurologic involvement. Other clinical features associated with IFAP syndrome consist of short stature, dysmorphic face, intestinal renal, cardiac and vertebral abnormalities, and cleft hands have been reported. Skin histopathology is non-specific [12-15].
IFAP syndrome results from missense mutations in the membranebound transcription factor protease site 2 (MBTPS2) gene. This gene encodes a zinc metalloproteinase intramembrane essential for cholesterol homeostasis and response of the endoplasmic reticulum to stress [3].
The therapeutic goal is essentially a multipronged approach that aimed to repair and protect the skin barrier, reduce microbial colonization and immune regulation to greater therapeutic efficacy, and improve quality of life of these patients. Follicular hyperkeratosis can be treated using topical keratolytics, urea preparations, and emollients. Oral retinoids at a dosage of 0.3 to 1 mg/kg/day have previously been used with moderate response. Photophobia and seizures may improve spontaneously, intensive lubrication of the ocular surface remains the mainstay of therapy, although sometimes antiseizure drugs are required [16]. For our case, the treatment was essentially based on urea emollients, local corticosteroids. Life expectancy in patients with IFAP syndrome can vary from death in the neonatal period to normal surviving. Cardiopulmonary complications remain the major cause of death [17].
IFAP syndrome is a rare syndrome. Its diagnosis is pratically clinical. The prognosis is mainly determined by cutaneous and extracutaneous manifestations. We have reported new observation of a boy non consanguineous parents whose clinic and genetic study were the keys to diagnosis, with a good improvement under topical treatment.