Author(s): Elharrouni Alaoui Aicha*, Elloudi Sara, Douhi Zakia, Baybay Hanane and Mernissi FZ
Poikilodermatous mycosis fungoides (PMF) is a rare clinical variant of early-stage MF with peculiar histological features. Poikiloderma occurs in many different clinical conditions, which makes a diagnosis procedure more complicated. PMF belongs to a group of MF variants with low risk of disease progression.We report 3 cases presented with mottled skin aspect of erythema, poikilodermatous patches (hypopigmentation, hyperpigmentation, atrophy, and telangiectasia) on more than 50-80% of the body. Based on clinical, histopathological, and immunohistochemical findings, we established the diagnosis of PMF.
Mycosis Fungoides is a primary epidermotropic cutaneous T-cell lymphoma. There has been an emergence of many clinicopathological and immunohistochemical variants of mycosis fungoides, sometimes atypical and misleading. We report 3 cases of a poikilodermic variant of mycosis fungoides.
A 71-year-old patient with hypertension, ACFA, dilated mitral stenosis and aortic valve replacement, consulting for diffuse skin lesions that had been evolving for 12 years. The clinical examination revealed erythematous plaques of variable size and shape, well limited, atrophic in places, with telangiectasias with a pigmented and reticular center giving the appearance of poïkilodermic lesions. These lesions were located the trunk and limbs occupying a skin surface estimated at 50% (Figure 1& 2). The biopsy revealed a histological and immunohistochemical profile of mycosis fungoides. The patient was classified IB and she was treated with topical steroids and methotrexate.
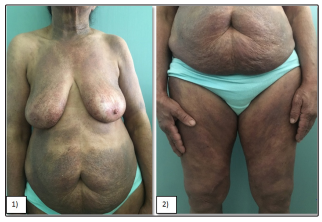
Figure 1 & 2: Female patient with predominant (50%)poikilodermatous lesions of mycosis fungoides.
A 58-year-old diabetic patient on insulin consulted for diffuse dyschromic pruriginous lesions on the entire body that had been evolving for 6 years. The dermatological examination revealed hypo and hyperpigmented plaques with irregular borders, atrophic in places scattered with telangiectasias giving a poikilodermic aspect, distributed all over the body with a cutaneous surface estimated at 70% (figure3-4). The histological and immunohistochemical study of a skin biopsy was in favor of a poikilodermic mycosis fungoides. The patient was classified as stage IB and she was successfully treated with PUVA therapy plus retinoid (Re-PUVA).
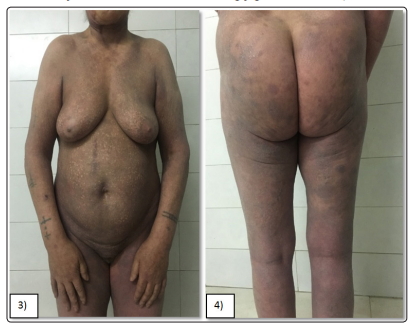
Figure 3&4: Female patient with predominant (70%) poikilodermatous lesions of mycosis fungoides.
A 59-year-old man had widespread poikilodermatous patches for 30 years. Physical examination revealed erythematous and scaly patches that were typically atrophic, telangiectatic, and intermingled with mottled hyperpigmentation and hypopigmentation, distributed all over the body with a cutaneous surface estimated at 90% (figure5-6). Skin biopsy specimen showed epidermotropism of atypical lymphocytes. There were vacuolar alterations of the basal layer, scattered melanophages, intermittent fibrosis of the collagen, and lymphocytic infiltration in the papillary dermis. These atypical lymphocytes were positive for CD3, CD4, and CD45RO but were negative for CD8, CD20, CD79. All these findings indicated the diagnosis of poikilodermatous mycosis fungoides (MF). Further detailed examination did not reveal any visceral or lymph node involvement. The patient was classified IB and she was treated with methotrexate and topical steroids.
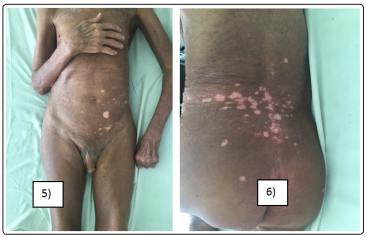
Figure 5&6: Mal patient with predominant (90%) poikilodermatous lesions of mycosis fungoides.
Mycosis fungoides represents the most common type of cutaneous T-cell lymphoma. It affects adults or the elderly. Three clinical stages can be recognized: one characterized by patches, one by patches and/or plaques, and one by patches, plaques, and/or tumours. In the early stages, preferential location is the buttocks and other sun-protected areas. Distinct clinical variants of MF have been reported and these may have a different clinical course when compared with classic MF, are less common and include granulomatous slack skin, pagetoid reticulosis, hypopigmented MF, poikilodermatous MF, and folliculotropic MF [1,2].
Poikilodermal MF, which is classically referred to as poikilo-derma vasculare atrophicans, is one of the most common variants of MF. In 1906 the pioneering American pediatrician Abraham Jacobi described a complex dermatologic disease by telangiectasia, pigmentation, and atrophy, which he subsequently termed poikiloderma vasculare atrophicans (PVA). It initially was considered a premalignant condition that would eventually progress to mycosis fungoides [3]. Nowadays poikiloderma vasculare atrophicans is recognized as a clinical variant of patch stage MF [4].
Poikilodermatous MF may be localized or diffuse ,is characterized clinically by “cigarette-paper” skin with the typical features of reticulate or mottled hyper- and hypopigmentation, atrophy, scaling, and telangiectasia, predominantly located on the breast, hips, and buttocks [5]. It may coexist with patches of classic MF in some patients ,may be asymptomatic or mildly pruritic [2].
Acquired poikiloderma may also appear as a feature of a number of conditions, including lupus erythematosus (LE), dermatomyositis (DM), poikiloderma of Civatte, radiation dermatitis, and topical corticosteroid overuse.
The histologic features may resemble those of classic patch or plaque stage MF whose Pautrier micro abscess sesare not usually present, but additional features include: epidermal atrophy, basal hydropic degeneration and associated interface dermatitis, pigment incontinence, and telangiectatic vessels [6]. In most cases, immunohistology shows a memory T-helper phenotype (CD3+, CD4+, CD45Ro+, CD8-, 45Ra-). CD30 and cytotoxic markers may be positive in late stages in tumors with large cell morphology [2,7,8]. Rarely, cases of MF are characterized by a CD8+ cytotoxic T-cell phenotype [2]. Nevertheless, the presence of this rare variant has neither prognostic significance nor therapeutic implications. The initial treatment is stage related.
The standard treatment for early lesions includes topical steroids, topical chemotherapy, PUVA, narrow-band UV-B, interferonα -2a, or oral retinoids. Combination therapies, which may reduce the toxicity of the single agents, are increasingly being used. Systemic chemotherapies and new immunologic agents, including denileukin diftitox, anti-CD52 antibody (alemtuzumab), and interleukin-12, are used in advanced stages with nodal or visceral involvement [9,10]. Overall there is a good response to phototherapy and the overall prognosis appears favorable.
Pokilodermic mycosis fungoides represents a clinicopathological entity that is rare and distinct from the classical form. Overall it has a good response to phototherapy and the prognosis seems favorable.