Author(s): Stanislaw R Burzynski*, Gregory S Burzynski, Tomasz Janicki and Samuel Beenken
Medulloblastomas arise in the posterior fossa, primarily in the region of the fourth ventricle. Recurrent medulloblastomas are high-risk and have a bad prognosis. Objectives: the case of a male child with recurrent, disseminated medulloblastoma is presented to detail the efficacy of ANP therapy (Antineoplaston A10 {Atengenal} and Antineoplaston AS2-1 {Astugenal}) in the treatment of high-risk medulloblastoma. This child was treated at the Burzynski Clinic (BC), as a Special Exception, according to Protocol BT-12, “Phase II Study of Antineoplastons A10 and AS2-1 in Children with Primitive Neuroectodermal Tumors”, with intravenous (IV) ANP therapy via a subclavian catheter and infusion pump. Tumor response was measured by sequential magnetic resonance imaging (MRI) of the brain with gadolinium enhancement. Findings: This child initially underwent tumor resection, placement of ventricular shunts, and chemotherapy, all performed elsewhere. Baseline MRI at the BC revealed recurrent, disseminated, and enhancing disease measuring 9.10 cm2 in total. IV ANP therapy began in March 2001 and ended in December 2002 after a complete response (CR) was achieved based on MRI criteria. Subsequently, the child received oral Antineoplastons as maintenance therapy, which were discontinued after eight months. At last follow-up, > 21 years and eight months since diagnosis and > 21 years and five months since the start of IV ANP therapy, the patient was well and showing no evidence of tumor recurrence. Conclusions: The utilization of ANP therapy to cure a patient with recurrent medulloblastoma is presented. We conclude that ANP therapy is an attractive therapeutic option for children with medulloblastoma.
Embryonal tumors, while biologically heterogeneous, tend to disseminate along cerebrospinal fluid (CSF) pathways. Their diagnosis is based primarily on histological and immunohistological features. More recently, genetic studies are being utilized to subclassify embryonal tumors for the purposes of risk stratification and treatment planning [1-4]. Approximately five percent of children with medulloblastoma have germline mutations, which puts them at risk of developing other cancers [5, 6].
Medulloblastomas arise in the posterior fossa, with nearly 80% located in the region of the fourth ventricle. In children, early symptoms are often due to blockage of the flow of CSF and resultant hydrocephalus. Common signs and symptoms include headaches, nausea and/or vomiting, lethargy, ataxia, and nystagmus. Signs and symptoms of medulloblastoma in infants may include lethargy, delay in psychomotor development, difficulty feeding, and increased head circumference [7].
On examination, one may see bulging of the anterior fontanel due to increased intracranial pressure while the eyes may deviate downward secondary to the loss of upward gaze, which is due to compression of the tectum of the midbrain [7].
Most medulloblastomas display some degree of anaplasia on histological examination. These features include increased nuclear size, marked cytological pleomorphism, numerous mitoses, and apoptotic bodies. [8, 9] If anaplasia is diffuse (50% - 80% of the tumor) one can consider the medulloblastoma to be anaplastic.
Histological findings of desmoplasia may indicate an improved outcome [10, 11]. However, patient age, the extent of disease at the time of diagnosis, histologic factors, and treatment received are currently being supplemented by the analysis of genetic variations in determining prognosis [12, 13]. In addition, genetic variations are being used to assign treatment in prospective studies [14].
Patient age, radiographical evaluation for disseminated disease, the amount of residual tissue after surgery, and CSF cytological examination have been used to project outcome [8]. Children less than 3 - 5 years of age with no dissemination and ≤ 1.5 cm2 of residual disease are considered “average risk” while thosedisseminated disease and/or > 1.5 cm2 of residual disease are considered “high risk” [15].
We present here the successful use of ANP therapy (Antineoplaston A10 {Atengenal} and Antineoplaston AS2-1 {Astugenal}) in the treatment of a one-year and seven-month-old male child with recurrent and disseminated medulloblastoma (high-risk) measuring 9.1 cm2 in total size (see below) [8].
The one-year and seven-month-old male child presented here was evaluated at the Burzynski Clinic (BC) on March 6, 2001. He was accompanied by his father and mother. The patient had headaches, slight weakness of the left upper and lower extremities, and a delay in motor skills, being unable to walk or crawl.
This child was in good health until December of 2000, when he lagged behind his developmental milestones, and was seen by his pediatrician. A magnetic resonance imaging (MRI) scan of the brain on December 19, 2000, demonstrated a midline cerebellar mass measuring 4.5 cm x 4.5 cm x 5.0 cm and extensive hydrocephalus, all of which were consistent with a medulloblastoma. Nodules on the cerebellar surface provided evidence of local tumor spread. On December 19, 2000, the child underwent a right frontal ventriculostomy and on December 22, 2000, he had a posterior fossa craniotomy with removal of the tumor. Histological examination of the resected tumor confirmed the diagnosis of medulloblastoma. A spinal fluid tap was negative for malignant cells. On January 1, 2001, the child developed GI bleeding and subsequently underwent laparotomy, ligation of a bleeding vessel in a duodenal ulcer, pyloroplasty, and gastrostomy. On January 2, 2001, an MRI scan of the brain demonstrated no evidence of residual tumor while MRI of the spine showed no evidence of CSF spread. On January 9, 2001, the child underwent removal of the right frontal ventriculostomy and placement of a left frontal ventriculostomy. On January 23, 2001, he again had placement of a right frontal ventriculostomy while the left frontal ventriculostomy catheter was removed. On February 12, 2001, the child began vincristine and cyclophosphamide chemotherapy, but only received one treatment before the patient was admitted to the hospital, on February 14, 2001, due to sepsis. He was treated with IV antibiotics. Chemotherapy was not resumed.
An MRI scan of the brain and spine, performed on March 2, 2001
(Figure 1), showed areas of enhancement in the right tentorium
and the right cerebellar hemisphere indicating tumor spread in the
brain. There was no evidence of CSF spread to the spine.
At the BC, on March 13, 2001, this patient began intravenous (IV)
ANP therapy, as a Special Exception (LPS = 50%, discontinued
chemotherapy), according to Protocol BT-12, a “Phase II Study
of Antineoplastons A10 and AS2-1 in Children with Primitive
Neuroectodermal Tumors.” In this single arm study, IV ANP
therapy was delivered every four hours via a subclavian catheter
and a programmable infusion pump.
The objectives of BT-12 were to 1) determine the efficacy of ANP
therapy in children with primitive neuroectodermal tumors as
determined by an objective response (OR) to therapy; 2) determine
OR utilizing MRI scans of the brain, which were performed every
8 weeks for the first two years, and then less frequently; and 3 )
determine the safety and tolerance of ANP therapy in this group
of patients
Eligibility criteria for BT-12 included 1) histologically confirmed
primitive neuroectodermal tumor; 2) evidence of persistent/
recurrent tumor as determined by gadolinium enhanced brain
MRI performed within 2 weeks of study enrollment; 3 ) Tumor size
≥ 5mm; 4) Age of 6 months to 17 years; 5) Lansky Performance
Status (LPS) ≥ 60%; and 6) Life expectancy ≥ 2 months.
Gadolinium enhanced MRI scans of the brain were used in the
diagnosis and follow-up of the medulloblastomas. T2-weighted,
T2-fluid attenuated inversion recovery (T2-FLAIR), T1 weighted
and T1-weighted contrast-enhanced images were obtained.
Medulloblastomas are gadolinium-enhancing, therefore sequential
T1-weighted contrast-enhanced images were utilized to determine
the effect of therapy [16].
As determined by MRI scan of the brain, the product of the two greatest perpendicular diameters of each measurable (≥ 5mm) and enhancing lesion was calculated. Tumor size was defined as the sum of these products. The response criteria were as follows: a complete response (CR) indicated complete disappearance of all enhancing tumor while a partial response (PR) indicated a 50% or greater reduction in total measurable and enhancing tumor size. CR and PR required a confirmatory brain MR scan performed at least four weeks after the initial OR. Progressive disease (PD) indicated a 25% or greater increase in total measurable and enhancing tumor size, or new measurable and enhancing disease, while stable disease (SD) did not meet the criteria for PR or PD [17].
Protocol BT-12 was conducted in accordance with the U.S. Code of Federal Regulations, Title 21, Parts 11, 50, 56 and 312; the Helsinki Declaration of 1975 (2000 revision); the Good Clinical Practices: Consolidated Guideline (E6), International Conference on Harmonization (ICH) and Guidance for Industry (FDA). By participating in this study protocol, the investigators agreed to provide access to all appropriate documents for monitoring, auditing, IRB review and review by any authorized regulatory agency. This Phase II study is described in Clinicaltrials.gov (CDR0000066492, NCT00003460).
Between April 1966 and January 2005, 13 children were accrued to BT-12 and treated at the BC. Twelve patients were evaluable while one was not evaluable due to technical problems with the follow-up MRI scans of the brain. Median age was 6.1 years (range: 1.0 to 12.2 years). Ten children were male while 3 were female. Among the eight children with medulloblastoma, one child obtained a CR, one child maintained SD, and six children developed PD.
As previously discussed, the child presented here was evaluated at the BC for recurrent medulloblastoma following resection of a primary posterior fossa tumor and placement of ventricular shunts. This child was treated at the BC, as a Special Exception (LPS = 50%, discontinued chemotherapy), according to Protocol BT-12, a “Phase II Study of Antineoplastons A10 and AS2-1 in Children with Primitive Neuroectodermal Tumors”. In this single arm study, IV ANP therapy was delivered every four hours via a subclavian catheter and a programmable infusion pump.
The starting dose of A10 for this child was 0.83 g/kg/d. It was gradually increased to 17.33 g/kg/d and then reduced to 13.23 g/ kg/d. His starting dose of AS2-1 was 0.22 g/kg/d. It was gradually increased to 0.46 g/kg/d and then reduced to 0.36 g/kg/d. Upon completion of IV ANP therapy and achievement of a CR, the child received oral Antineoplastons as maintenance therapy, which was discontinued after eight months.
On September 18, 2001, a six-month follow-up MRI scan of the brain showed a 59.1% decrease in size of the recurrent medulloblastoma, indicating a PR. On June 3, 2002, one-year and 3-month follow-up brain MRI scan showed no evidence of residual enhancing disease, indicating a CR (Figure 1). On March 21, 2005, MRI scan of the brain showed no evidence of residual enhancing disease, indicating an enduring CR (Figure 1). At last follow-up, August 30, 2022, the patient was doing very well and had received no other anti-tumor therapy. OS has been > 21 years and eight months since diagnosis and > 21 years and five months since the start of ANP therapy. All MRIs of the brain showing an OR were reviewed by a prominent outside neuroradiologist.
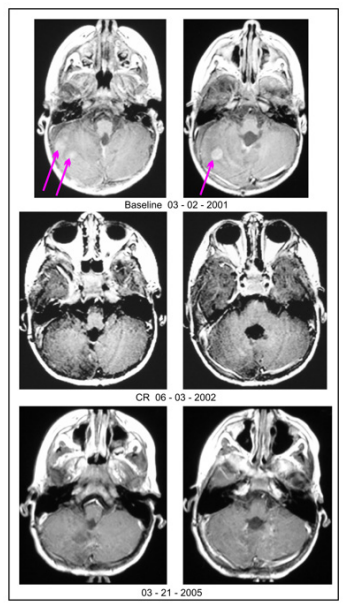
Figure 1: Axial images from MRI brain scans performed before, during, and after ANP therapy: March 2, 2001 - Baseline MRI showing recurrent and disseminated medulloblastoma (see arrows); June 3, 2002 - One-year and 3-month follow-up MRI showing no evidence of residual enhancing disease, indicating a CR; March 21, 2005 - Follow-up MRI showing no evidence of residual enhancing disease, indicating an enduring CR. ANP therapy = Antineoplaston A10 (Atengenal) and Antineoplaston AS2-1 (Astugenal); MRI = magnetic resonance imaging; PR = partial response; CR = complete response.
Adverse events (AEs) were graded according to the Common Terminology Criteria for Adverse Events Version 3.0 (CTCAE v.3). Ten patients accrued to BT-12 experienced a serious adverse event (SAE). The child presented here developed two SAEs thought to be due to ANP therapy, hypernatremia and hypocalcemia, which occurred during a period of somnolence brought on by aspiration pneumonia. He recovered fully from the pneumonia, the somnolence, the hypernatremia, and the hypocalcemia.
In the 2021 World Health Organization (WHO) classification, medulloblastoma is characterized according to histological and genetic findings [18]. If there is no genetic testing, the classification is medulloblastoma, histologically defined.
The treatment of younger children with newly diagnosed
medulloblastoma is not fully standardized. The use of different
drug regimens and the variable use of craniospinal and local
boost radiation therapy (RT), in children ≥ 3 years of age, makes
comparison of studies difficult. However certain treatment
guidelines can be emphasized.
Total surgical resection of the tumor is the optimal initial treatment.
Subsequently, adjuvant chemotherapy is frequently utilized, being
more frequently based on genetic analysis, and includes drugs such
as cyclophosphamide, etoposide, cisplatin, and vincristine, with or
without concomitant high-dose intravenous and/or intraventricular
methotrexate [19-21].
In infants and young children with desmoplastic medulloblastoma, medulloblastoma with extensive nodularity (MBEN), and tumors with sonic-hedgehog (SHH) pathway mutation signaling, high dose systemic chemotherapy and intraventricular methotrexate or high-dose systemic chemotherapy followed by stem cell rescue, without radiation, is effective treatment [22]. In children with localized disease after chemotherapy, focal RT does not improve survival [23]. Long-term survivors treated with chemotherapy are at a high risk of later hearing loss, cardiac complications, and secondary neoplasms. In infants and young children with a non-desmoplastic, non-MBEN, and non-SHH pathway mutation signaling medulloblastoma, high dose systemic chemotherapy and intraventricular methotrexate or high-dose systemic chemotherapy followed by stem cell rescue, without radiation, is not effective treatment for these children, who have a much worse prognosis [24]. There is no standard therapy in recurrent medulloblastoma.
We have presented the use of IV ANP therapy in a child diagnosed at age one-year and four-months with a recurrent medulloblastoma. He was treated with IV ANP therapy and achieved a CR after one-year and three-months of therapy, which, at the time of his last follow-up, had persisted for > 21 years and five months since the start of IV ANP therapy.
Antineoplaston (ANP) research began in 1967, when significant deficiencies were noticed in the peptide content of the serum of patients with cancer compared with healthy persons. Initially ANP were isolated from the blood and later from urine [25]. Subsequent studies of the isolated ANP demonstrated that Antineoplaston A-10 and Antineoplaston AS2-1 were the most active ANPs. The chemical name of Antineoplaston A-10 is 3-phenylacetylamino2,6-piperidinedione. It consists of the cyclic form of L-glutamine connected by a peptide bond to phenylacetyl residue. When given orally, Antineoplaston A10 resists the attack of gastric enzymes. In the small intestine, under alkaline conditions, 30% is digested into phenylacetylglutamine (PG) and phenylacetylisoglutaminate (isoPG) in a ratio of approximately 4:1. The mixture of synthetic PG and isoPG in a 4:1 ratio, dissolved in sterile water constitutes Antineoplaston A10 intravenous (IV) injection. Further metabolism of Antineoplaston A10 results in phenylacetate (PN). Bothmetabolites PG and PN have anticancer activity. The mixture of PN and PG in a 4:1 ratio, dissolved in sterile water constitutes Antineoplaston AS2-1 IV injection [26].
ANP therapy’s mechanism of action differs from that of RT or cytotoxic chemotherapy. Growth of normal cells is controlled by cell cycle progression genes (oncogenes) and by cell cycle arrest genes (tumor suppressor genes). In cancer, alteration of these control genes in malignant cells favors aggressive cell proliferation. Evidence suggests that ANP therapy affects 204 mutated genes in the malignant genome and functions as a “molecular switch” which “turns on” tumor-suppressor genes and “turns off” oncogenes [27, 28]. Hence, the antineoplastic action of ANP therapy in medulloblastoma involves restoration of cell cycle control, induction of programmed cell death, and interference with cancer cell metabolism and nuclear transport.
We have presented here the case of a male child with a high-risk recurrent and disseminated medulloblastoma who obtained a CR with ANP therapy, which indicates that ANP therapy may be an effective therapeutic option for children with medulloblastoma. Multiple Phase II clinical studies of ANP therapy in a variety of low- and high-grade brain tumors under the Burzynski Research Institute’s (BRI’s) IND # 43,742 have now been completed and numerous articles have been published [29-72]. Based on the CR described here, we propose a multi-institutional Phase II study of ANP therapy for children with medulloblastoma.
The authors express their appreciation to Carolyn Powers for preparation of the manuscript and to Ramiro Rivera, Mohamed Khan, Jennifer Pineda and Adam Golunski for their involvement.